Азербайджан, країна, що швидко розвивається в Кавказькому регіоні, представляє багатообіцяючу можливість для міжнародних компаній-виробників медичних виробів, які прагнуть розширити свою присутність на ринку. З населенням близько 10 мільйонів і зростаючим сектором охорони здоров’я в Азербайджані спостерігається підвищений попит на інноваційні та високоякісні медичні вироби.

Управління регуляторним середовищем для медичних виробів в Азербайджані може бути складним завданням для міжнародних менеджерів з регуляторних питань, які не знайомі зі специфічними вимогами та процедурами країни.
Цей всеосяжний посібник має на меті надати міжнародним менеджерам з регуляторних питань необхідну інформацію та знання для впевненого виходу на азербайджанський ринок та оптимізації їхніх зусиль з реєстрації медичних виробів.
Ми сподіваємося надати вам чітке розуміння:
- Основні регуляторні органи та їх ролі в реєстрації медичних виробів
- Законодавча база, що регулює медичні вироби в Азербайджані
- Система класифікації медичних виробів та її значення для реєстрації
- Загальна та спрощена процедури реєстрації та вимоги до них
- Післяреєстраційні зобов’язання, включаючи маркування, пильність та постмаркетинговий нагляд
- Правила імпорту та експорту медичних виробів
- Рекомендації для успішного виходу на ринок та дотримання вимог
Регуляторні органи та законодавча база в Азербайджані
Міністерство охорони здоров’я Азербайджанської Республіки
Міністерство охорони здоров’я Азербайджанської Республіки є основним регуляторним органом, відповідальним за нагляд за сектором охорони здоров’я, включаючи регулювання медичних виробів. Основні обов’язки Міністерства включають
- Розробка та реалізація національної політики та стратегій, пов’язаних з охороною здоров’я та медичними виробами
- Видача ліцензій на діяльність, пов’язану з медичними виробами, таку як виробництво, імпорт та дистрибуція
- нагляд за реєстрацією, контролем якості та моніторингом безпеки медичних виробів
- Співпраця з міжнародними організаціями та іноземними регуляторними органами з питань, пов’язаних з медичними виробами.
Міністерство охорони здоров’я складається з декількох департаментів та підпорядкованих органів, які відіграють певну роль у регулюванні медичних виробів. Найбільш важливими для менеджерів з міжнародних регуляторних питань є наступні департаменти:
- Департамент фармацевтичної політики та регулювання: Відповідає за розробку та впровадження політики, нормативних актів та настанов, пов’язаних з медичними виробами та фармацевтичними препаратами.
- Департамент ліцензування та сертифікації: займається ліцензуванням діяльності, пов’язаної з медичними виробами, та сертифікацією фахівців з медичних виробів.
- Департамент міжнародних зв’язків: Координує співпрацю Міністерства з міжнародними організаціями та іноземними регуляторними органами у сфері медичних виробів.
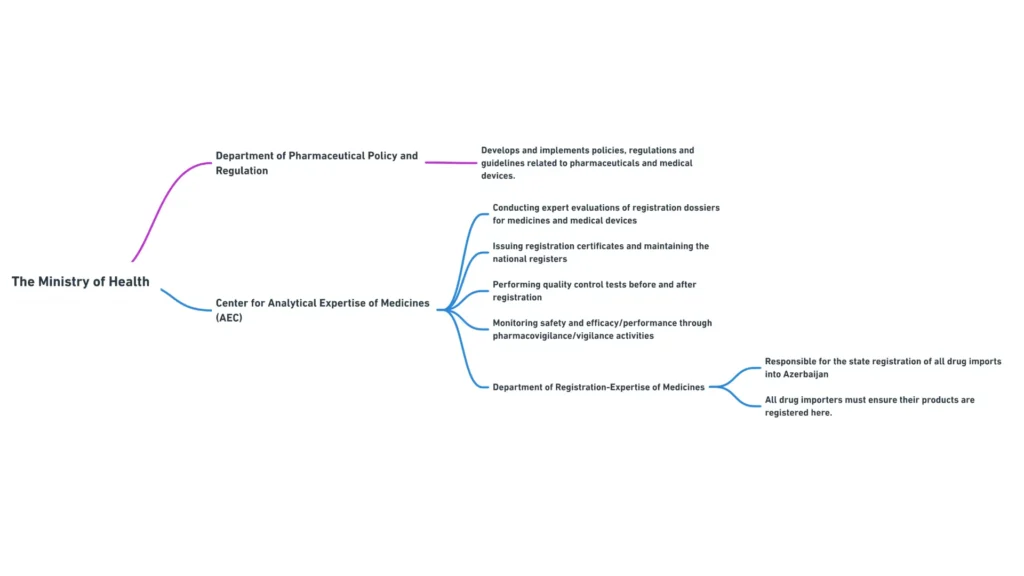
Структура Міністерства охорони здоров’я Азербайджану та його департаментів
Центр аналітичної експертизи лікарських засобів
Центр аналітичної експертизи лікарських засобів, підпорядкований Міністерству охорони здоров’я, відіграє вирішальну роль у реєстрації та контролі якості медичних виробів в Азербайджані.
Основні функції Центру включають
- проведення експертизи реєстраційних досьє на медичні вироби
- Видача реєстраційних посвідчень та ведення національного реєстру медичних виробів
- Проведення випробувань з контролю якості медичних виробів до та після реєстрації
- Моніторинг безпеки та ефективності зареєстрованих медичних виробів за допомогою заходів з нагляду.
Центр аналітичної експертизи лікарських засобів є основною контактною особою для менеджерів з міжнародних регуляторних питань під час процесу реєстрації.
Підтримання чіткої комунікації з Центром та оперативне реагування на будь-які запити або прохання про надання додаткової інформації має вирішальне значення для забезпечення безперебійного процесу реєстрації.
Відповідне законодавство та нормативні акти
Нормативно-правова база, що регулює обіг медичних виробів в Азербайджані, складається із законів, указів і постанов, які встановлюють вимоги та процедури реєстрації, виробництва, імпорту та дистрибуції медичних виробів.
До найбільш важливих законодавчих актів, з якими слід ознайомитися менеджерам з міжнародних регуляторних питань, належать такі:
- Закон “Про лікарські засоби” (2006): основний закон, що регулює медичні вироби та фармацевтичну продукцію в Азербайджані. Він охоплює такі аспекти, як реєстрація, виробництво, контроль якості та нагляд. З моменту прийняття закону до нього було внесено кілька поправок з метою приведення його у відповідність до міжнародних стандартів та найкращих практик.
- Постанова № 108 “Про затвердження Правил державної реєстрації лікарських засобів” (2007): Викладає детальні вимоги та процедури для реєстрації медичних виробів та фармацевтичної продукції, включаючи зміст реєстраційного досьє, строки та збори.
- Постанова № 137 “Про правила проведення експертизи лікарських засобів” (2007): Визначає процедури та критерії експертної оцінки реєстраційних досьє Центром аналітичної експертизи лікарських засобів.
- Постанова № 137 “Про правила імпорту та експорту лікарських засобів” (2007): встановлює вимоги та процедури імпорту та експорту медичних виробів і фармацевтичної продукції, включаючи ліцензування, митне оформлення та документацію.
На додаток до цих ключових законодавчих актів, менеджери з міжнародних регуляторних питань також повинні бути в курсі будь-яких змін, додаткових нормативних актів або керівних документів, виданих Міністерством охорони здоров’я або Центром аналітичної експертизи лікарських засобів.
Регулярне відвідування офіційних веб-сайтів та інформаційних бюлетенів цих органів допоможе забезпечити відповідність найсучаснішим вимогам.
Класифікація медичних виробів в Азербайджані
В Азербайджані діє система класифікації медичних виробів на основі оцінки ризиків, яка значною мірою гармонізована з системою класифікації Європейського Союзу відповідно до Регламенту щодо медичних виробів (MDR). Класифікація медичного виробу визначає застосовні процедури оцінки відповідності та рівень регуляторного нагляду, необхідний для реєстрації.
Правила та критерії класифікації
Медичні вироби поділяються на чотири категорії залежно від їхнього призначення, тривалості використання, інвазивності та потенційних ризиків для пацієнта або користувача.
Правила класифікації враховують такі фактори, як
- Спосіб дії пристрою та джерело енергії
- Ступінь і тривалість контакту з тілом людини
- Потенційна можливість заподіяння шкоди або травм у разі несправності або неправильного використання
- Залежність пристрою від джерела живлення або програмного забезпечення
- Використання пристрою в поєднанні з іншими пристроями або речовинами
Чотири класифікаційні категорії:
- Клас I: Пристрої з низьким рівнем ризику, які не контактують з пацієнтом або мають лише короткий контакт з неушкодженою шкірою (наприклад, нестерильні бинти, оглядові рукавички та допоміжні засоби для ходьби).
- Клас IIa: Пристрої низького та середнього ризику, які контактують з пошкодженою шкірою, вводяться в отвори тіла або використовуються для короткочасних інвазивних процедур (наприклад, відсмоктувачі, слухові апарати та пломбувальні матеріали для зубів).
- Клас IIb: Пристрої середнього та високого ризику, які використовуються для довготривалих інвазивних процедур, імплантуються або можуть спричинити травми у разі несправності (наприклад, апарати штучної вентиляції легень, ортопедичні імплантати та інфузійні насоси).
- Клас III: Пристрої підвищеного ризику, які використовуються для виконання критично важливих функцій, контактують з центральною нервовою системою або системою кровообігу, або містять речовини людського або тваринного походження (наприклад, серцеві клапани, неврологічні імплантати і нитки, що розсмоктуються).
На додаток до цих загальних правил класифікації, існують спеціальні правила для певних типів пристроїв, таких як протизаплідні засоби, дезінфікуючі засоби та радіологічні пристрої.
Ці спеціальні правила можуть призвести до більш високої класифікації, ніж та, що була б присвоєна на основі лише загальних правил.
Приклади класифікацій пристроїв та їх вплив на реєстрацію
Щоб проілюструвати систему класифікації та її вплив на реєстрацію, розглянемо наступні приклади:
- Нестерильні марлеві пов’язки (Клас I): Нестерильні марлеві пов’язки відносяться до класу I. Як вироби з низьким рівнем ризику, що мають лише короткий контакт з неушкодженою шкірою, нестерильні марлеві пов’язки відносяться до класу I. Виробник повинен підготувати технічний файл, що демонструє відповідність основним вимогам, але не зобов’язаний залучати нотифікований орган до процесу оцінки відповідності. Реєстраційне досьє на пристрій класу I є відносно простим і може бути швидко опрацьоване органами влади.
- Слухові апарати (клас IIa): Слухові апарати класифікуються як пристрої класу IIa, оскільки вони використовуються в отворах тіла (вушний канал) і можуть завдати шкоди, якщо вони несправні. Виробник повинен підготувати більш розширений технічний файл і залучити нотифікований орган до процесу оцінки відповідності. Реєстраційне досьє на вироби класу IIa вимагає більш детальної документації і може зайняти більше часу, ніж на вироби класу I.
- Ортопедичні імплантати (клас IIb): Ортопедичні імплантати, такі як протези кульшового або колінного суглоба, класифікуються як пристрої класу IIb через їхню довготривалу інвазивність і потенційну небезпеку серйозних травм у разі відмови. Виробник повинен підготувати вичерпний технічний файл, включаючи клінічні дані, і пройти більш сувору оцінку відповідності нотифікованим органом. Реєстраційне досьє на виріб класу IIb є складним і може вимагати додаткової експертизи з боку уповноважених органів, що призводить до збільшення часу на його обробку.
- Серцеві клапани (Клас III): Серцеві клапани відносяться до Класу III, оскільки вони є критично важливими для функціонування системи кровообігу і можуть призвести до серйозних наслідків у разі їхньої несправності.
Виробник повинен підготувати обширний технічний файл, що включає суттєві клінічні дані, і пройти ретельну оцінку відповідності нотифікованим органом. Реєстраційне досьє на пристрій класу III є дуже складним і може вимагати додаткової експертизи та консультацій з клінічними фахівцями, що призводить до найдовшого часу обробки серед усіх класів пристроїв.
Точно класифікувавши свої пристрої та підготувавши відповідну документацію, виробники можуть спростити процес реєстрації та уникнути непотрібних затримок або відмов.
Процедури та терміни реєстрації в Азербайджані
Щоб зареєструвати медичний виріб в Азербайджані, менеджери з міжнародних регуляторних питань повинні дотримуватися відповідної процедури реєстрації та подати повне досьє до Центру аналітичної експертизи лікарських засобів.
Процес реєстрації складається з двох основних процедур: загальної процедури реєстрації та спрощеної процедури реєстрації.
Загальна процедура реєстрації медичних виробів в Азербайджані
Загальна процедура реєстрації є стандартним шляхом реєстрації медичних виробів в Азербайджані. Ця процедура передбачає ретельну оцінку якості, безпеки та ефективності пристрою Центром аналітичної експертизи лікарських засобів.
Основні етапи та вимоги загальної процедури реєстрації наведені нижче:
- Подання досьє: Заявник повинен подати повне реєстраційне досьє до Центру аналітичної експертизи лікарських засобів. Досьє повинно включати наступні документи:
- Адміністративні документи (наприклад, форма заявки, довіреність, сертифікати)
- Технічна документація (наприклад, опис пристрою, призначення, інформація про дизайн та виробництво, аналіз ризиків, клінічна оцінка)
- Маркування та пакувальні матеріали (наприклад, інструкції із застосування, етикетки, макети)
- Досьє подається в паперовому та електронному вигляді разом з необхідним реєстраційним збором. Адміністративна частина досьє повинна бути азербайджанською або російською мовою, в той час як технічна документація може бути азербайджанською, російською або англійською мовами.
- Попередній розгляд: Після отримання досьє Центр аналітичної експертизи лікарських засобів проводить попередню експертизу, щоб переконатися, що заявка є повною і відповідає формальним вимогам. Якщо будуть виявлені будь-які недоліки, заявник буде повідомлений про це і йому буде надано певний термін для їх усунення.
- Спеціалізована експертиза: Після того, як досьє проходить попередній розгляд, воно проходить спеціалізовану експертизу, яка передбачає детальну оцінку якості, безпеки та ефективності пристрою. Цей етап включає в себе
- Оцінку технічної документації
- Лабораторний аналіз зразків пристроїв, за необхідності
- Аналіз клінічних даних та документації з управління ризиками
- На цьому етапі Центр аналітичної експертизи лікарських засобів може запросити у заявника додаткову інформацію або роз’яснення. Дуже важливо оперативно та ретельно відповідати на ці запити, щоб уникнути затримок у процесі реєстрації.
- Рішення та видача свідоцтва: За результатами спеціалізованої експертизи Центр аналітичної експертизи лікарських засобів приймає рішення про схвалення або відхилення заявки на реєстрацію. У разі позитивного рішення заявник отримає реєстраційне посвідчення, дійсне протягом п’яти років. Зареєстрований виріб також буде внесено до національного реєстру медичних виробів.
Загальна процедура реєстрації зазвичай займає від 30 до 45 днів від дати подання досьє до видачі реєстраційного посвідчення.
Однак цей термін може змінюватися залежно від складності пристрою, якості поданого досьє та завантаженості Центру аналітичної експертизи лікарських засобів.
Спрощена процедура реєстрації медичних виробів в Азербайджані
Азербайджан пропонує спрощену процедуру реєстрації медичних виробів, які вже були зареєстровані в країнах із суворими регуляторними органами або отримали маркування CE відповідно до Регламенту Європейського Союзу щодо медичних виробів (MDR).
Ця процедура має на меті спростити процес реєстрації виробів, які пройшли сувору оцінку визнаних регуляторних органів.
Щоб мати право на спрощену процедуру реєстрації, медичний виріб повинен відповідати одному з наступних критеріїв:
- Виріб зареєстровано в країнах із суворими регуляторними органами (наприклад, ЄС, США, Канада, Японія, Австралія)
- Пристрій отримав маркування CE відповідно до Регламенту ЄС щодо медичних виробів (MDR)
За спрощеною процедурою реєстрації заявник повинен подати скорочений пакет документів, зокрема:
- Заява на реєстрацію та довіреність
- Свідоцтво про реєстрацію або сертифікат СЕ з референтної країни
- Інструкція для застосування та матеріали для маркування азербайджанською мовою
Центр аналітичної експертизи лікарських засобів розгляне подані документи та прийме рішення про реєстрацію протягом 30 днів.
У разі позитивного рішення заявник отримає реєстраційне посвідчення, дійсне протягом такого ж терміну, як і реєстраційне посвідчення або сертифікат СЕ референтної країни, але не більше ніж п’ять років.
Важливо зазначити, що спрощена процедура реєстрації не звільняє вироби від вимоги демонструвати якість, безпеку та ефективність. Центр аналітичної експертизи лікарських засобів може запросити додаткову інформацію або роз’яснення в процесі експертизи.
Відмова в реєстрації та процес оскарження
У деяких випадках Центр аналітичної експертизи лікарських засобів може відмовити в реєстрації медичного виробу. Найпоширенішими підставами для відмови є
- Недостатні або неадекватні дані для демонстрації якості, безпеки або продуктивності пристрою
- невідповідність відповідним стандартам або регуляторним вимогам
- Спотворення або фальсифікація інформації в реєстраційному досьє
- Неусунення недоліків або ненадання додаткової інформації на вимогу органів влади у встановлений термін.
У разі відмови в реєстрації заявник отримає письмове повідомлення із зазначенням причин відмови. Заявник може оскаржити рішення протягом 30 днів шляхом подання письмового запиту до Міністерства охорони здоров’я.
Апеляція буде розглянута експертною комісією вищого рівня, яка прийме остаточне рішення щодо реєстраційної заявки.
Післяреєстраційні зобов’язання в Азербайджані
Після успішної реєстрації медичного виробу в Азербайджані виробники та уповноважені представники повинні дотримуватися різних післяреєстраційних зобов’язань, щоб підтримувати реєстрацію пристрою і забезпечувати його постійну безпеку та ефективність.
Вимоги до маркування та пакування в Азербайджані
Усі медичні вироби, що продаються в Азербайджані, повинні відповідати певним вимогам до маркування та пакування, щоб забезпечити доступ медичних працівників і пацієнтів до точної та повної інформації про виріб.
Вимоги до мови та змісту в Азербайджані
Маркування та пакувальні матеріали для медичних виробів повинні бути азербайджанською мовою.
На етикетці та в інструкції з використання повинна міститися наступна інформація:
- Назва та модель пристрою
- Назва та адреса виробника
- Призначення та показання до застосування
- Протипоказання, застереження та запобіжні заходи
- Інструкція із застосування, зберігання та утилізації
- Номер партії або серійний номер
- Термін придатності або дата виготовлення
- Статус стерильності та метод стерилізації, якщо застосовується
- маркування CE або реєстраційний номер, якщо застосовно
Крім азербайджанської мови, маркування та пакувальні матеріали можуть також надаватися російською, англійською або турецькою мовами.
Специфічні вимоги до певних категорій виробів
До певних категорій медичних виробів можуть висуватися додаткові вимоги щодо маркування та пакування. Наприклад:
- Стерильні пристрої повинні містити інформацію про метод стерилізації та термін придатності стерильності
- Пристрої, що містять лікарські речовини, повинні містити інформацію про склад і дозування речовини
- Пристрої, що випромінюють іонізуюче випромінювання, повинні містити попередження та запобіжні заходи, пов’язані з радіаційним впливом
Процес затвердження змін до маркування та пакування
Будь-які зміни до затвердженого маркування або пакувальних матеріалів повинні бути подані до Центру аналітичної експертизи лікарських засобів на розгляд та затвердження до їх впровадження.
Заявник повинен надати детальний опис запропонованих змін та їх обґрунтування, а також оновлені зразки маркування та пакування.
Центр аналітичної експертизи лікарських засобів розгляне запропоновані зміни та прийме рішення про їх затвердження протягом 30 днів.
Якщо зміни будуть визнані прийнятними, заявник отримає повідомлення про затвердження і може приступити до впровадження оновленого маркування та упаковки.
Пильність та повідомлення про побічні реакції в Азербайджані
Виробники та уповноважені представники несуть відповідальність за постійний моніторинг безпеки та ефективності своїх медичних виробів і повідомляють про будь-які несприятливі події або інциденти до Центру аналітичної експертизи лікарських засобів.
Вимоги до створення системи пильності
Виробники повинні створити та підтримувати систему фармаконагляду, яка відповідає вимогам Закону “Про лікарські засоби” та відповідних нормативно-правових актів.
Ключові елементи системи пильності включають
- Процедури збору, оцінки та повідомлення про несприятливі події та інциденти
- Призначена контактна особа з питань пильності, відповідальна за комунікацію з органами влади
- Ведення звітності та документації з питань пильності
- Проведення аналізу тенденцій та оцінки ризиків на основі даних пильності
Терміни та процедури звітування
Виробники та уповноважені представники повинні повідомляти до Центру аналітичної експертизи лікарських засобів про наступні типи подій:
- Серйозні побічні реакції: протягом 15 календарних днів з моменту, коли стало відомо про подію
- Інші несприятливі події та інциденти: протягом 30 календарних днів після того, як стало відомо про подію
- Коригувальні дії з безпеки на місцях (наприклад, відкликання, модифікації): до або одночасно з реалізацією дії
Звіти слід подавати за визначеними формами та каналами, наданими Центром аналітичної експертизи лікарських засобів. Звіти повинні містити детальний опис події, відповідного(их) пристрою(ів), а також заходів, вжитих або запланованих виробником для вирішення проблеми.
Співпраця з регуляторними органами у випадку інцидентів або відкликань
У разі виникнення серйозного інциденту або необхідності проведення коригувальних заходів з безпеки на місцях, виробники та уповноважені представники повинні в повній мірі співпрацювати з Центром аналітичної експертизи лікарських засобів та іншими відповідними органами.
Ця співпраця може включати
- Надання додаткової інформації та документації за запитом
- Проведення розслідувань та аналізу першопричин
- Впровадження коригувальних та профілактичних дій
- Спілкування з медичними працівниками та пацієнтами
- Сприяння відкликанню або модифікації уражених пристроїв
Невиконання вимог щодо пильності та повідомлень про несприятливі події може призвести до покарань, таких як штрафи, призупинення або анулювання реєстрації пристрою, а у важких випадках – до кримінальної відповідальності.
Постмаркетинговий нагляд та клінічне спостереження
Виробники зобов’язані здійснювати постмаркетинговий нагляд та клінічне спостереження для забезпечення постійної безпеки та ефективності своїх медичних виробів протягом усього їхнього життєвого циклу.
Зобов’язання щодо здійснення постмаркетингового нагляду
Зобов’язання щодо постмаркетингового нагляду включають
- Збір та аналіз даних про безпеку та експлуатаційні характеристики пристрою з різних джерел (наприклад, звіти про пильність, скарги споживачів, наукова література)
- Проведення періодичних оглядів безпеки та оновлення документації з управління ризиками пристрою
- Впровадження коригувальних та запобіжних дій для вирішення виявлених проблем або ризиків
- Ведення записів і документації з постмаркетингового нагляду
Періодичні звіти з безпеки (ПЗБ)
Виробники повинні подавати періодичні звіти з безпеки (ПЗБ) до Центру аналітичної експертизи лікарських засобів згідно з наступним графіком:
- Пристрої III класу: щорічно
- Пристрої класу IIb: кожні 2 роки
- Пристрої класу IIa: кожні 3 роки
- Пристрої класу I: не вимагається
ПУР повинен надавати всебічний огляд безпеки та ефективності пристрою, включаючи узагальнення даних щодо пильності, постмаркетингового нагляду, а також будь-яких змін або оновлень в документації з управління ризиками для пристрою.
Клінічні наглядові дослідження для медичних виробів високого ризику в Азербайджані
Для пристроїв високого ризику (наприклад, класу III та деяких пристроїв класу IIb) виробники можуть бути зобов’язані проводити постмаркетингові клінічні дослідження з метою подальшої оцінки безпеки та ефективності пристрою в реальних умовах.
Необхідність проведення клінічних досліджень буде визначатися Центром аналітичної експертизи лікарських засобів в кожному конкретному випадку з урахуванням таких факторів, як
- Новизна та складність пристрою
- Наявність та якість домаркетингових клінічних даних
- Потенційні ризики, пов’язані з використанням пристрою
- Цільова популяція пацієнтів та передбачуване використання
Виробники повинні подати протокол клінічного спостереження до Центру аналітичної експертизи лікарських засобів на розгляд та затвердження перед початком дослідження.
Результати дослідження повинні бути подані до уповноважених органів у визначений термін і можуть бути використані для оновлення маркування пристрою, документації з управління ризиками або реєстраційного статусу.
Поновлення реєстрації медичних виробів та зміна реєстрації в Азербайджані
Реєстрації медичних виробів в Азербайджані є дійсними протягом обмеженого періоду часу і повинні поновлюватися, щоб зберегти дозвіл на маркетинг пристрою. Крім того, будь-які суттєві зміни або модифікації зареєстрованого виробу повинні бути повідомлені або схвалені Центром аналітичної експертизи лікарських засобів.
Термін дії реєстраційних свідоцтв на медичні вироби
Реєстраційні свідоцтва на медичні вироби в Азербайджані, як правило, дійсні протягом п’яти років з дати видачі. Однак для виробів, зареєстрованих за спрощеною процедурою на підставі іноземної реєстрації або сертифіката CE, термін дії може бути коротшим і відповідати даті закінчення терміну дії відповідної іноземної реєстрації або сертифіката CE.
Процес подання заяви на перереєстрацію та вимоги
Для перереєстрації медичного виробу виробники або уповноважені представники повинні подати заяву на перереєстрацію до Центру аналітичної експертизи лікарських засобів щонайменше за 90 днів до закінчення терміну дії поточної реєстрації.
Заява на перереєстрацію повинна містити
- Оновлену технічну документацію, включаючи будь-які зміни або модифікації, внесені в пристрій з моменту первинної реєстрації
- Актуальне маркування та пакувальні матеріали
- Дані пильності та постмаркетингового нагляду
- Підтвердження сплати збору за перереєстрацію
Центр аналітичної експертизи лікарських засобів розгляне заяву на перереєстрацію та прийме рішення про перереєстрацію протягом 30 днів. Якщо заявка буде схвалена, виробник отримає нове реєстраційне посвідчення, дійсне протягом наступних п’яти років.
Повідомлення та затвердження змін до зареєстрованих медичних виробів
Виробники повинні повідомляти Центр аналітичної експертизи лікарських засобів про будь-які суттєві зміни або модифікації зареєстрованого медичного виробу.
Типи змін, які потребують повідомлення або затвердження, включають
- Зміни в конструкції, складі або процесі виробництва пристрою
- Зміни в призначенні пристрою
Правила імпорту та експорту в Азербайджані
Менеджери з міжнародних регуляторних питань також повинні бути обізнані з правилами імпорту та експорту медичних виробів в Азербайджані, щоб забезпечити їх дотримання та сприяти безперешкодній транскордонній торгівлі.
Ліцензійні вимоги до імпортерів та експортерів медичних виробів
Компанії, що займаються імпортом або експортом медичних виробів в Азербайджані, повинні отримати відповідні ліцензії від Міністерства охорони здоров’я.
Ліцензія імпортера
Для імпорту медичних виробів до Азербайджану компанії повинні отримати ліцензію імпортера. Основні вимоги для отримання ліцензії імпортера включають
- Реєстрація юридичної особи в Азербайджані
- Наявність відповідних приміщень та обладнання для зберігання та поводження з медичними виробами
- Наявність кваліфікованого персоналу, включаючи відповідальну особу за контроль якості та дотримання нормативних вимог
- Впровадження системи управління якістю, яка відповідає відповідним стандартам і нормам.
Заява на отримання ліцензії імпортера подається до Міністерства охорони здоров’я разом із супровідними документами, такими як свідоцтво про реєстрацію компанії, план приміщення та кваліфікація персоналу. Міністерство розгляне заяву та прийме рішення протягом 30 днів.
Ліцензовані імпортери несуть відповідальність за те, щоб імпортовані медичні вироби були зареєстровані в Азербайджані, відповідали чинним нормативно-правовим актам, а також зберігалися і використовувалися відповідно до належної практики дистрибуції.
Ліцензія експортера
Компанії, які експортують медичні вироби з Азербайджану, повинні отримати ліцензію експортера. Вимоги до ліцензії експортера подібні до вимог до ліцензії імпортера, з додатковою вимогою мати дійсну ліцензію на виробництво або контракт з ліцензованим виробником.
Процес подання заявки на отримання ліцензії експортера також подібний до процесу подання заявки на отримання ліцензії імпортера: Міністерство охорони здоров’я розглядає заявку та приймає рішення протягом 30 днів.
Ліцензовані експортери несуть відповідальність за те, щоб експортовані медичні вироби відповідали нормативним вимогам країни призначення і супроводжувалися необхідною документацією, такою як сертифікати походження та відповідності.
Процедури митного оформлення та документація в Азербайджані
При імпорті або експорті медичних виробів компанії повинні дотримуватися процедур митного оформлення та надавати необхідну документацію.
Необхідні документи для імпорту та експорту
Для імпорту та експорту медичних виробів до Азербайджану зазвичай потрібні наступні документи:
- Комерційний інвойс
- Пакувальний лист
- Сертифікат походження
- Сертифікат відповідності або реєстрації
- Товаросупровідні документи (наприклад, коносамент, авіанакладна)
- Ліцензія на імпорт або експорт
- Інші документи, які вимагають митні органи
Важливо переконатися, що всі документи є точними, повними та легалізованими або апостильованими, якщо це необхідно.
Співпраця з митними органами
Компанії, що імпортують або експортують медичні вироби, повинні співпрацювати з митними органами та надавати будь-яку додаткову інформацію або документацію, що запитується в процесі митного оформлення.
Це може включати
- Надання зразків пристроїв для перевірки або тестування
- Роз’яснення призначення або класифікації пристроїв
- Демонстрація відповідності чинним нормам і стандартам
Відмова від співпраці з митними органами або ненадання необхідної документації може призвести до затримок, штрафів або відхилення вантажу.
Особливі міркування щодо пристроїв, які містять підконтрольні речовини або потребують управління холодовим ланцюгом
На медичні вироби, які містять підконтрольні речовини (наприклад, наркотики, психотропні речовини) або потребують дотримання холодового ланцюга (наприклад, термочутливі пристрої), поширюються додаткові вимоги щодо імпорту та експорту.
Для пристроїв, що містять підконтрольні речовини, імпортери та експортери повинні отримати спеціальні дозволи від Міністерства охорони здоров’я та дотримуватися правил поводження з підконтрольними речовинами та їх транспортування.
Для пристроїв, що вимагають дотримання холодового ланцюга, імпортери та експортери повинні забезпечити зберігання та транспортування пристроїв за відповідних температурних умов, а також надати докази моніторингу та контролю температури під час митного оформлення.
Рекомендації для міжнародних компаній
Щоб успішно орієнтуватися на азербайджанському ринку медичних виробів, міжнародні менеджери з регуляторних питань повинні враховувати наступні рекомендації:
Важливість залучення місцевих регуляторних експертів та партнерів
Залучення місцевих регуляторних експертів і партнерів, таких як Delta Medical, може надати неоціненну допомогу в розумінні та дотриманні азербайджанського законодавства щодо медичних виробів. Місцеві експерти можуть допомогти:
- Тлумачити та застосовувати відповідні закони та нормативні акти
- Готувати та подавати реєстраційні досьє та інші необхідні документи
- Комунікація з регуляторними органами та відповіді на запити
- Відстежувати регуляторні зміни та оновлювати стратегії комплаєнсу
Обираючи місцевих партнерів, міжнародні компанії повинні враховувати такі фактори, як досвід, експертиза, репутація та здатність партнера надавати всебічну підтримку протягом усього життєвого циклу продукту.
Останні думки щодо реєстрації медичних виробів в Азербайджані
Для успішного завершення процедур реєстрації медичних виробів в Азербайджані менеджери з міжнародних регуляторних питань повинні врахувати наступні рекомендації:
- Залучайте місцевих регуляторних експертів та партнерів: Співпраця з місцевими регуляторними експертами та партнерами, такими як Delta Medical, може допомогти міжнародним компаніям зрозуміти та виконати вимоги Азербайджану, уникнути поширених помилок та прискорити процес реєстрації.
- Плануйте заздалегідь і виділяйте достатньо часу для реєстрації: Процес реєстрації в Азербайджані може бути тривалим і складним, особливо для інноваційних продуктів. Щоб уникнути затримок з виходом на ринок, міжнародні компанії повинні планувати заздалегідь і виділити достатньо часу на підготовку досьє, подання та розгляд, щоб уникнути затримок з виходом на ринок.
- Надавати пріоритет продуктам з високим ринковим потенціалом: Щоб максимізувати віддачу від інвестицій, міжнародні компанії повинні надавати пріоритет реєстрації продуктів з високим ринковим потенціалом в Азербайджані, беручи до уваги такі фактори, як поширеність захворювань, незадоволені медичні потреби та конкуренція.
- Забезпечити дотримання післяреєстраційних вимог: Міжнародні компанії повинні створити надійні системи та процеси фармаконагляду для дотримання післяреєстраційних вимог в Азербайджані. Регулярний перегляд і оновлення цих систем може допомогти уникнути штрафних санкцій і забезпечити безперебійну роботу бізнесу.
- Будьте в курсі регуляторних змін та оновлень: Регуляторне середовище Азербайджану постійно розвивається, періодично з’являються нові нормативно-правові акти та інструкції. Менеджери з міжнародних регуляторних питань повинні бути в курсі цих змін і відповідно адаптувати свої стратегії та процеси.
Дотримуючись цих рекомендацій та тісно співпрацюючи з місцевими партнерами, міжнародні компанії можуть успішно управляти регуляторним ринком Азербайджану та сприяти покращенню доступу пацієнтів до безпечних, ефективних та якісних медичних виробів.


