Azerbaijan, a rapidly developing country in the Caucasus region, presents a promising opportunity for international medical device companies seeking to expand their market presence. With a population of approximately 10 million and a growing healthcare sector, Azerbaijan has seen an increasing demand for innovative and high-quality medical devices.

Managing the regulatory landscape for medical devices in Azerbaijan can be challenging for international regulatory affairs managers who are unfamiliar with the country’s specific requirements and procedures.
This comprehensive guide aims to provide international regulatory affairs managers with the necessary information and insights to confidently approach the Azerbaijani market and streamline their device registration efforts.
We hope to provide you with a clear understanding of:
- The main regulatory authorities and their roles in medical device registration
- The legal framework governing medical devices in Azerbaijan
- The classification system for medical devices and its implications for registration
- The general and simplified registration procedures and their requirements
- Post-registration obligations, including labeling, vigilance, and post-market surveillance
- Import and export regulations for medical devices
- Recommendations for successful market entry and compliance
Regulatory Authorities and Legal Framework in Azerbaijan
Ministry of Health of the Republic of Azerbaijan
The Ministry of Health of the Republic of Azerbaijan is the primary regulatory authority responsible for overseeing the healthcare sector, including the regulation of medical devices. The Ministry’s key responsibilities include:
- Developing and implementing national policies and strategies related to healthcare and medical devices
- Issuing licenses for medical device-related activities, such as manufacturing, importing, and distributing
- Overseeing the registration, quality control, and safety monitoring of medical devices
- Collaborating with international organizations and foreign regulatory authorities on matters related to medical devices
The Ministry of Health comprises several departments and subordinate bodies that play specific roles in medical device regulation. The most relevant departments for international regulatory affairs managers include:
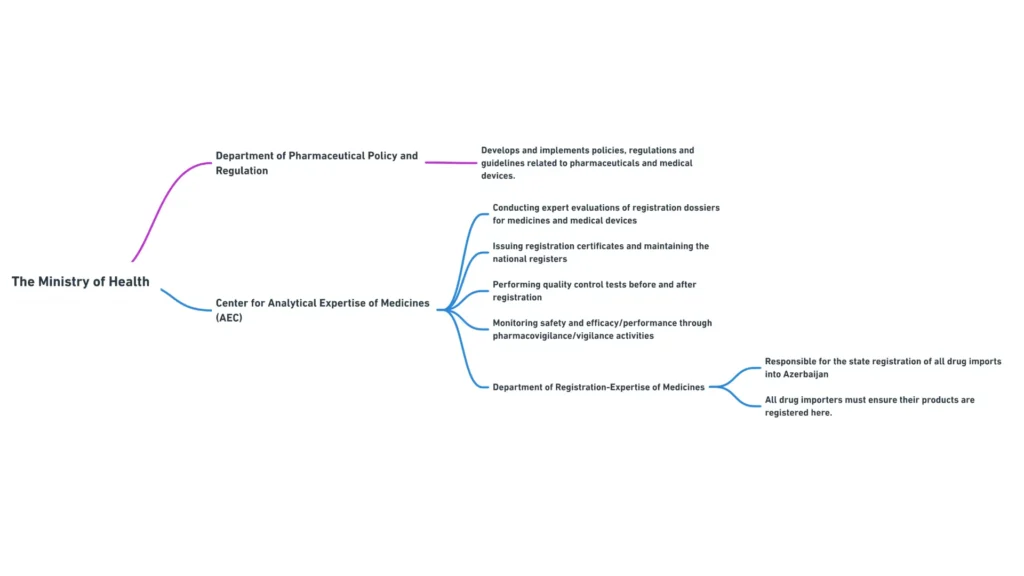
- Department of Pharmaceutical Policy and Regulation: Responsible for developing and implementing policies, regulations, and guidelines related to medical devices and pharmaceuticals.
- Department of Licensing and Certification: Handles the licensing of medical device-related activities and the certification of medical device professionals.
- Department of International Relations: Coordinates the Ministry’s cooperation with international organizations and foreign regulatory authorities in the field of medical devices.
Structure of Azerbaijan MOH and Departments
Center for Analytical Expertise of Medicines
The Center for Analytical Expertise of Medicines, a subordinate body of the Ministry of Health, plays a crucial role in the registration and quality control of medical devices in Azerbaijan.
The Center’s main functions include:
- Conducting expert evaluations of registration dossiers for medical devices
- Issuing registration certificates and maintaining the national register of medical devices
- Performing quality control tests on medical devices before and after registration
- Monitoring the safety and performance of registered medical devices through vigilance activities
The Center for Analytical Expertise of Medicines serves as the primary point of contact for international regulatory affairs managers during the registration process.
Maintaining clear communication with the Center and promptly addressing any queries or requests for additional information is crucial for ensuring a smooth registration process.
Relevant Legislation and Regulations
The legal framework governing medical devices in Azerbaijan consists of laws, decrees, and regulations that set out the requirements and procedures for the registration, manufacture, import, and distribution of medical devices.
The most important legislation for international regulatory affairs managers to be aware of includes:
- Law on Medicines (2006): The primary law regulating medical devices and pharmaceutical products in Azerbaijan. It covers aspects such as registration, manufacturing, quality control, and vigilance. The law has undergone several amendments since its adoption to align with international standards and best practices.
- Decree No. 108 on the Rules for State Registration of Medicines (2007): Outlines the detailed requirements and procedures for the registration of medical devices and pharmaceutical products, including the content of registration dossiers, timelines, and fees.
- Decree No. 137 on the Rules for Conducting Expertise of Medicines (2007): Specifies the procedures and criteria for the expert evaluation of registration dossiers by the Center for Analytical Expertise of Medicines.
- Resolution No. 137 on the Rules for Importing and Exporting Medicines (2007): Sets out the requirements and procedures for importing and exporting medical devices and pharmaceutical products, including licensing, customs clearance, and documentation.
In addition to these key pieces of legislation, international regulatory affairs managers should also stay informed about any amendments, supplementary regulations, or guidance documents issued by the Ministry of Health or the Center for Analytical Expertise of Medicines.
Regularly consulting the official websites and newsletters of these authorities can help ensure compliance with the most up-to-date requirements.
Medical Device Classification in Azerbaijan
Azerbaijan follows a risk-based classification system for medical devices, which is largely harmonized with the European Union’s classification system under the Medical Device Regulation (MDR). The classification of a medical device determines the applicable conformity assessment procedures and the level of regulatory oversight required for registration.
Classification Rules and Criteria
Medical devices are classified into four categories based on their intended purpose, duration of use, invasiveness, and potential risks to the patient or user.
The classification rules take into account factors such as:
- The device’s mode of action and energy source
- The degree and duration of contact with the human body
- The potential for harm or injury in case of malfunction or misuse
- The device’s reliance on a power source or software
- The device’s use in combination with other devices or substances
The four classification categories are:
- Class I: Low-risk devices that do not come into contact with the patient or only have brief contact with intact skin (e.g., non-sterile bandages, examination gloves, and walking aids).
- Class IIa: Low-to-medium risk devices that come into contact with injured skin, are introduced into body orifices, or are used for short-term invasive procedures (e.g., suction equipment, hearing aids, and dental filling materials).
- Class IIb: Medium-to-high risk devices that are used for long-term invasive procedures, are implantable, or have the potential to cause injury in case of malfunction (e.g., ventilators, orthopedic implants, and infusion pumps).
- Class III: High-risk devices that are used for critical functions, are in contact with the central nervous system or circulatory system, or contain substances of human or animal origin (e.g., heart valves, neurological implants, and absorbable sutures).
In addition to these general classification rules, there are special rules for certain types of devices, such as contraceptive devices, disinfectants, and radiological devices.
These special rules may result in a higher classification than would be assigned based on the general rules alone.
Examples of Device Classifications and Their Implications for Registration
To illustrate the classification system and its implications for registration, consider the following examples:
- Non-sterile gauze dressings (Class I): As a low-risk device with only brief contact with intact skin, non-sterile gauze dressings are classified as Class I. The manufacturer must prepare a technical file demonstrating compliance with the essential requirements but does not need to involve a notified body in the conformity assessment process. The registration dossier for a Class I device is relatively simple and can be processed quickly by the authorities.
- Hearing aids (Class IIa): Hearing aids are classified as Class IIa devices because they are used in body orifices (the ear canal) and have the potential to cause harm if they malfunction. The manufacturer must prepare a more extensive technical file and involve a notified body in the conformity assessment process. The registration dossier for a Class IIa device requires more detailed documentation and may take longer to process than a Class I device.
- Orthopedic implants (Class IIb): Orthopedic implants, such as hip or knee prostheses, are classified as Class IIb devices due to their long-term invasiveness and the potential for serious injury in case of failure. The manufacturer must prepare a comprehensive technical file, including clinical data, and undergo a more rigorous conformity assessment by a notified body. The registration dossier for a Class IIb device is complex and may require additional expert review by the authorities, resulting in longer processing times.
- Heart valves (Class III): Heart valves are classified as Class III devices because they are critical for the functioning of the circulatory system and have the potential for severe consequences in case of malfunction.
The manufacturer must prepare an extensive technical file, including substantial clinical data, and undergo a thorough conformity assessment by a notified body. The registration dossier for a Class III device is highly complex and may require additional expert review and consultation with clinical specialists, resulting in the longest processing times among all device classes.
By accurately classifying their devices and preparing the appropriate documentation, manufacturers can streamline the registration process and avoid unnecessary delays or rejections.
Registration Procedures and Timelines in Azerbaijan
To register a medical device in Azerbaijan, international regulatory affairs managers must follow the appropriate registration procedure and submit a comprehensive dossier to the Center for Analytical Expertise of Medicines.
The registration process consists of two main procedures: the general registration procedure and the simplified registration procedure.
General Medical Device Registration Procedure in Azerbaijan
The general registration procedure is the standard pathway for registering medical devices in Azerbaijan. This procedure involves a thorough evaluation of the device’s quality, safety, and performance by the Center for Analytical Expertise of Medicines.
The key steps and requirements of the general registration procedure are as follows:
- Dossier Submission: The applicant must submit a complete registration dossier to the Center for Analytical Expertise of Medicines. The dossier should include the following documents:
- Administrative documents (e.g., application form, power of attorney, certificates)
- Technical documentation (e.g., device description, intended use, design and manufacturing information, risk analysis, clinical evaluation)
- Labeling and packaging materials (e.g., instructions for use, labels, mock-ups)
- The dossier must be submitted in both hard copy and electronic format, along with the required registration fee. The administrative part of the dossier should be in Azerbaijani or Russian, while the technical documentation can be in Azerbaijani, Russian, or English.
- Preliminary Review: Upon receiving the dossier, the Center for Analytical Expertise of Medicines conducts a preliminary review to ensure that the application is complete and meets the formal requirements. If any deficiencies are identified, the applicant will be notified and given a specified timeframe to address them.
- Specialized Expertise: Once the dossier passes the preliminary review, it undergoes a specialized expertise, which involves a detailed evaluation of the device’s quality, safety, and performance. This stage includes:
- Assessment of the technical documentation
- Laboratory analysis of device samples, if required
- Review of clinical data and risk management documentation
- During this stage, the Center for Analytical Expertise of Medicines may request additional information or clarification from the applicant. It is crucial to respond promptly and thoroughly to these requests to avoid delays in the registration process.
- Decision and Certificate Issuance: Based on the outcome of the specialized expertise, the Center for Analytical Expertise of Medicines makes a decision on whether to approve or reject the registration application. If approved, the applicant will receive a registration certificate valid for five years. The registered device will also be included in the national register of medical devices.
The general registration procedure typically takes between 30 to 45 days from the date of dossier submission to the issuance of the registration certificate.
However, this timeline may vary depending on the complexity of the device, the quality of the submitted dossier, and the workload of the Center for Analytical Expertise of Medicines.
Simplified Medical Device Registration Procedure in Azerbaijan
Azerbaijan offers a simplified registration procedure for medical devices that have already been registered in countries with stringent regulatory authorities or have obtained CE marking under the European Union’s Medical Device Regulation (MDR).
This procedure aims to streamline the registration process for devices that have undergone rigorous evaluation by recognized regulatory bodies.
To be eligible for the simplified registration procedure, a medical device must meet one of the following criteria:
- The device is registered in countries with stringent regulatory authorities (e.g., EU, USA, Canada, Japan, Australia)
- The device has obtained CE marking under the EU’s Medical Device Regulation (MDR)
Under the simplified registration procedure, the applicant must submit a reduced set of documents, including:
- Application form and power of attorney
- Certificate of registration or CE certificate from the reference country
- Instructions for use and labeling materials in Azerbaijani
The Center for Analytical Expertise of Medicines will review the submitted documents and make a decision on the registration within 30 days.
If approved, the applicant will receive a registration certificate valid for the same period as the registration or CE certificate from the reference country, up to a maximum of five years.
It is important to note that the simplified registration procedure does not exempt devices from the requirement to demonstrate quality, safety, and performance. The Center for Analytical Expertise of Medicines may still request additional information or clarification during the review process.
Refusal of Registration and Appeal Process
In some cases, the Center for Analytical Expertise of Medicines may refuse to register a medical device. The most common grounds for refusal include:
- Insufficient or inadequate data to demonstrate the device’s quality, safety, or performance
- Non-compliance with relevant standards or regulatory requirements
- Misrepresentation or falsification of information in the registration dossier
- Failure to address deficiencies or provide additional information requested by the authorities within the specified timeframe
If a registration application is refused, the applicant will receive a written notification outlining the reasons for the refusal. The applicant may appeal the decision within 30 days by submitting a written request to the Ministry of Health.
The appeal will be reviewed by a higher-level expert committee, which will make a final decision on the registration application.
Post-Registration Obligations in Azerbaijan
Once a medical device has been successfully registered in Azerbaijan, manufacturers and authorized representatives must comply with various post-registration obligations to maintain the device’s registration and ensure its ongoing safety and performance.
Labeling and Packaging Requirements in Azerbaijan
All medical devices marketed in Azerbaijan must comply with specific labeling and packaging requirements to ensure that healthcare professionals and patients have access to accurate and complete information about the device.
Language and Content Requirements in Azerbaijan
Labeling and packaging materials for medical devices must be in the Azerbaijani language.
The following information must be included on the label and instructions for use:
- Device name and model
- Manufacturer’s name and address
- Intended use and indications
- Contraindications, warnings, and precautions
- Instructions for use, storage, and disposal
- Lot or serial number
- Expiration date or manufacturing date
- Sterility status and sterilization method, if applicable
- CE marking or registration number, if applicable
In addition to Azerbaijani, labeling and packaging materials may also be provided in Russian, English, or Turkish.
Specific Requirements for Certain Device Categories
Certain categories of medical devices may be subject to additional labeling and packaging requirements. For example:
- Sterile devices must include information on the sterilization method and expiration date of sterility
- Devices containing medicinal substances must include information on the composition and dosage of the substance
- Devices emitting ionizing radiation must include warnings and precautions related to radiation exposure
Approval Process for Labeling and Packaging Changes
Any changes to the approved labeling or packaging materials must be submitted to the Center for Analytical Expertise of Medicines for review and approval prior to implementation.
The applicant must provide a detailed description of the proposed changes and their rationale, along with updated labeling and packaging samples.
The Center for Analytical Expertise of Medicines will review the proposed changes and make a decision on their approval within 30 days.
If the changes are deemed acceptable, the applicant will receive a notification of approval and may proceed with implementing the updated labeling and packaging.
Vigilance and Adverse Event Reporting in Azerbaijan
Manufacturers and authorized representatives are responsible for continuously monitoring the safety and performance of their medical devices and reporting any adverse events or incidents to the Center for Analytical Expertise of Medicines.
Requirements for Establishing a Vigilance System
Manufacturers must establish and maintain a vigilance system that complies with the requirements set out in the Law on Medicines and relevant regulations.
The key elements of a vigilance system include:
- Procedures for collecting, evaluating, and reporting adverse events and incidents
- Designated vigilance contact person responsible for communicating with the authorities
- Maintenance of vigilance records and documentation
- Conducting trend analysis and risk assessments based on vigilance data
Reporting Timelines and Procedures
Manufacturers and authorized representatives must report the following types of events to the Center for Analytical Expertise of Medicines:
- Serious adverse events: within 15 calendar days of becoming aware of the event
- Other adverse events and incidents: within 30 calendar days of becoming aware of the event
- Field safety corrective actions (e.g., recalls, modifications): prior to or simultaneously with the implementation of the action
Reports should be submitted using the designated forms and channels provided by the Center for Analytical Expertise of Medicines. The reports must include a detailed description of the event, the affected device(s), and the actions taken or planned by the manufacturer to address the issue.
Cooperation with Regulatory Authorities in Case of Incidents or Recalls
In the event of a serious incident or the need for a field safety corrective action, manufacturers and authorized representatives must cooperate fully with the Center for Analytical Expertise of Medicines and other relevant authorities.
This cooperation may include:
- Providing additional information and documentation upon request
- Conducting investigations and root cause analyses
- Implementing corrective and preventive actions
- Communicating with healthcare professionals and patients
- Facilitating the recall or modification of affected devices
Failure to comply with vigilance and adverse event reporting requirements may result in penalties, such as fines, suspension or revocation of the device’s registration, or criminal liability in severe cases.
Post-Market Surveillance and Clinical Follow-Up
Manufacturers are obligated to conduct post-market surveillance and clinical follow-up activities to ensure the ongoing safety and performance of their medical devices throughout their lifecycle.
Obligations for Conducting Post-Market Surveillance
Post-market surveillance obligations include:
- Collecting and analyzing data on the device’s safety and performance from various sources (e.g., vigilance reports, customer complaints, scientific literature)
- Conducting periodic safety reviews and updating the device’s risk management documentation
- Implementing corrective and preventive actions to address identified issues or risks
- Maintaining post-market surveillance records and documentation
Periodic Safety Update Reports (PSURs)
Manufacturers must submit periodic safety update reports (PSURs) to the Center for Analytical Expertise of Medicines according to the following schedule:
- Class III devices: annually
- Class IIb devices: every 2 years
- Class IIa devices: every 3 years
- Class I devices: not required
PSURs should provide a comprehensive overview of the device’s safety and performance, including a summary of vigilance data, post-market surveillance activities, and any changes or updates to the device’s risk management documentation.
Clinical Follow-Up Studies for High-Risk Devices in Azerbaijan
For high-risk devices (e.g., Class III and certain Class IIb devices), manufacturers may be required to conduct post-market clinical follow-up studies to further assess the device’s safety and performance in real-world settings.
The need for clinical follow-up studies will be determined by the Center for Analytical Expertise of Medicines on a case-by-case basis, taking into account factors such as:
- The novelty and complexity of the device
- The availability and quality of pre-market clinical data
- The potential risks associated with the device’s use
- The target patient population and intended use
Manufacturers must submit a clinical follow-up study protocol to the Center for Analytical Expertise of Medicines for review and approval prior to initiating the study.
The study results must be submitted to the authorities within the specified timeframe and may be used to update the device’s labeling, risk management documentation, or registration status.
Renewal of Medical Devices and Variation of Registration in Azerbaijan
Medical device registrations in Azerbaijan are valid for a limited period and must be renewed to maintain the device’s marketing authorization. Additionally, any significant changes or variations to the registered device must be notified to or approved by the Center for Analytical Expertise of Medicines.
Validity Period of Medical Device Registration Certificates
Medical device registration certificates in Azerbaijan are typically valid for five years from the date of issuance. However, for devices registered through the simplified registration procedure based on a foreign registration or CE certificate, the validity period may be shorter and aligned with the expiration date of the underlying foreign registration or CE certificate.
Renewal Application Process and Requirements
To renew a medical device registration, manufacturers or authorized representatives must submit a renewal application to the Center for Analytical Expertise of Medicines at least 90 days before the expiration of the current registration.
The renewal application should include:
- Updated technical documentation, including any changes or modifications made to the device since the initial registration
- Current labeling and packaging materials
- Vigilance and post-market surveillance data
- Proof of payment of the renewal fee
The Center for Analytical Expertise of Medicines will review the renewal application and make a decision on the renewal within 30 days. If the application is approved, the manufacturer will receive a new registration certificate valid for another five years.
Notification and Approval of Changes to Registered Medical Devices
Manufacturers must notify the Center for Analytical Expertise of Medicines of any significant changes or variations to a registered medical device.
The types of changes that require notification or approval include:
- Changes to the device’s design, composition, or manufacturing process
- Changes to the device’s intended use
Import and Export Regulations in Azerbaijan
International regulatory affairs managers must also be aware of the import and export regulations governing medical devices in Azerbaijan to ensure compliance and facilitate smooth cross-border trade.
Licensing Requirements for Medical Device Importers and Exporters
Companies engaged in the import or export of medical devices in Azerbaijan must obtain the appropriate licenses from the Ministry of Health.
Importer License
To import medical devices into Azerbaijan, companies must obtain an importer license. The key requirements for obtaining an importer license include:
- Registering a legal entity in Azerbaijan
- Having suitable premises and facilities for the storage and handling of medical devices
- Employing qualified personnel, including a responsible person for quality control and regulatory compliance
- Implementing a quality management system that complies with relevant standards and regulations
The importer license application must be submitted to the Ministry of Health, along with supporting documents such as the company’s registration certificate, premises layout, and staff qualifications. The Ministry will review the application and make a decision within 30 days.
Licensed importers are responsible for ensuring that imported medical devices are registered in Azerbaijan, comply with applicable regulations, and are stored and handled in accordance with good distribution practices.
Exporter License
Companies that export medical devices from Azerbaijan must obtain an exporter license. The requirements for an exporter license are similar to those for an importer license, with the additional requirement of having a valid manufacturing license or contract with a licensed manufacturer.
The exporter license application process is also similar to that of the importer license, with the Ministry of Health reviewing the application and making a decision within 30 days.
Licensed exporters are responsible for ensuring that exported medical devices comply with the regulations of the destination country and are accompanied by the necessary documentation, such as certificates of origin and conformity.
Customs Clearance Procedures and Documentation in Azerbaijan
When importing or exporting medical devices, companies must comply with the customs clearance procedures and provide the required documentation.
Required Documents for Import and Export
The following documents are typically required for the import and export of medical devices in Azerbaijan:
- Commercial invoice
- Packing list
- Certificate of origin
- Certificate of conformity or registration
- Shipping documents (e.g., bill of lading, air waybill)
- Import or export license
- Other documents as required by the customs authorities
It is important to ensure that all documents are accurate, complete, and legalized or apostilled as necessary.
Cooperation with Customs Authorities
Companies importing or exporting medical devices must cooperate with the customs authorities and provide any additional information or documentation requested during the clearance process.
This may include:
- Providing samples of the devices for inspection or testing
- Clarifying the intended use or classification of the devices
- Demonstrating compliance with applicable regulations and standards
Failure to cooperate with the customs authorities or provide the required documentation may result in delays, penalties, or the rejection of the shipment.
Special Considerations for Devices Containing Controlled Substances or Requiring Cold Chain Management
Medical devices that contain controlled substances (e.g., narcotics, psychotropic substances) or require cold chain management (e.g., temperature-sensitive devices) are subject to additional import and export requirements.
For devices containing controlled substances, importers and exporters must obtain special permits from the Ministry of Health and comply with the regulations governing the handling and transportation of controlled substances.
For devices requiring cold chain management, importers and exporters must ensure that the devices are stored and transported under the appropriate temperature conditions and provide evidence of temperature monitoring and control during customs clearance.
Recommendations for International Companies
To successfully navigate the Azerbaijani medical device market, international regulatory affairs managers should consider the following recommendations:
Importance of Engaging Local Regulatory Experts and Partners
Engaging local regulatory experts and partners, such as Delta Medical, can provide invaluable assistance in understanding and complying with Azerbaijani medical device regulations. Local experts can help:
- Interpret and apply the relevant laws and regulations
- Prepare and submit registration dossiers and other required documents
- Communicate with the regulatory authorities and respond to queries
- Monitor regulatory changes and update compliance strategies
When selecting local partners, international companies should consider factors such as the partner’s experience, expertise, reputation, and ability to provide comprehensive support throughout the product lifecycle.
Final Thoughts on Registering Medical Devices in Azerbaijan
To successfully complete the Azerbaijani medical device procedures, international regulatory affairs managers should consider the following recommendations:
- Engage Local Regulatory Experts and Partners: Working with local regulatory experts and partners, such as Delta Medical, can help international companies understand and comply with Azerbaijani requirements, avoid common pitfalls, and accelerate the registration process.
- Plan Ahead and Allow Sufficient Time for Registration: The registration process in Azerbaijan can be lengthy and complex, particularly for innovative products. International companies should plan ahead and allow sufficient time for dossier preparation, submission, and review to avoid delays in market entry.
- Prioritize Products with High Market Potential: To maximize the return on investment, international companies should prioritize the registration of products with high market potential in Azerbaijan, considering factors such as disease prevalence, unmet medical needs, and competition.
- Ensure Compliance with Post-Registration Requirements: International companies must establish robust pharmacovigilance systems and processes to comply with post-registration requirements in Azerbaijan. Regularly reviewing and updating these systems can help avoid penalties and maintain the smooth operation of the business.
- Stay Informed of Regulatory Changes and Updates: The Azerbaijani regulatory landscape is constantly evolving, with new regulations and guidelines being introduced periodically. International regulatory affairs managers should stay informed of these changes and adapt their strategies and processes accordingly.
By following these recommendations and working closely with local partners, international companies can successfully manage the Azerbaijani regulatory market and contribute to improving patient access to safe, effective, and high-quality medical devices.


