Фармаконадзор (PVG), включающий мониторинг безопасности лекарственных средств, крайне важен для фармацевтических компаний, чтобы обеспечить безопасность пациентов и соответствовать нормативным требованиям во всем мире.

Однако ориентироваться в сложных местных нормах, действующих в разных странах, может быть непросто.
Для международных компаний, выходящих на белорусский рынок или расширяющих свою деятельность, понимание системы PVG в стране необходимо, чтобы избежать таких последствий, как отзыв продукции, штрафы и репутационный ущерб от несоблюдения требований.
Нормативно-правовая база в Беларуси
Основные законодательные акты и руководства по фармаконадзору в Беларуси
Система фармаконадзора в Беларуси регулируется несколькими ключевыми законодательными актами и руководствами, включая:
- Закон о фармацевтической деятельности: Этот всеобъемлющий закон устанавливает правовые основы регулирования фармацевтической деятельности в Беларуси, включая положения, относящиеся к деятельности по фармаконадзору.
- Инструкция о порядке сообщения о неблагоприятных реакциях на фармацевтические препараты: В этом документе изложены конкретные требования и процессы сообщения о неблагоприятных событиях (НС) и серьезных неблагоприятных событиях (СНС), связанных с фармацевтической продукцией в Беларуси.
- Инструкция о порядке организации системы фармаконадзора: Данная инструкция содержит рекомендации по созданию и поддержанию эффективной системы фармаконадзора в фармацевтических компаниях, работающих в Беларуси.
- Принятие надлежащей практики фармаконадзора Евразийского экономического союза (ЕАЭС) : Беларусь, как государство-член ЕАЭС, приняла надлежащую практику фармаконадзора ЕАЭС, которая направлена на гармонизацию практики фармаконадзора в странах-членах союза. Соблюдение GVP ЕАЭС является обязательным для компаний, работающих в Беларуси.
Регулирующие органы в Беларуси
Основным регулирующим органом, ответственным за надзор за системой фармаконадзора в Беларуси, является Министерство здравоохранения. В рамках Министерства Центр экспертизы и испытаний в здравоохранении (ЦЭИТЗ) играет важнейшую роль в реализации и обеспечении соблюдения нормативных требований в области фармаконадзора.
CETH отвечает за:
- Получение, обработку и оценку отчетов о побочных реакциях на фармацевтические препараты
- Проведение инспекций и аудитов для обеспечения соблюдения правил PVG
- Предоставление рекомендаций и поддержки фармацевтическим компаниям по вопросам, связанным с PVG
- Сотрудничество с международными организациями и регуляторными органами для приведения в соответствие с лучшими мировыми практиками.
Мастер-файл системы фармаконадзора (PSMF) в Беларуси
В ГВП ЕАЭС введено понятие мастер-файла системы фармаконадзора (PSMF) — комплексного документа, в котором описывается система фармаконадзора компании и ее соответствие требованиям ГВП. Хотя PSMF ЕАЭС не является обязательным требованием в соответствии с национальным законодательством Беларуси, он считается дополнением к существующей документации.
Компании, работающие в Беларуси, должны быть готовы предоставить следующую информацию о своей ОУФП:
- Местонахождение ОФП: физическое и/или электронное место(а), где хранится и поддерживается ОФП.
- Представление PSMF: сроки и процедуры представления PSMF в CETH по запросу.
- Обновление PSMF: процессы регулярного обновления и поддержания PSMF для отражения изменений в системе фармаконадзора компании.
Таблица 1: Основные законодательные акты и руководства по фармаконадзору в Беларуси
| Законодательство/руководство | Описание |
| Закон о фармацевтических препаратах | Создает правовую базу для регулирования фармацевтической деятельности, включая положения о PVG |
| Инструкция по информированию о неблагоприятных реакциях | Описывает требования и процессы отчетности по АЭ и САЭ |
| Инструкция по организации системы PVG | Содержит руководство по созданию и поддержанию эффективной системы PVG |
| Надлежащая практика ЕАЭС в области фармаконадзора (GVP) | Гармонизированное руководство, принятое Беларусью как страной-членом ЕАЭС |

Отчетность о неблагоприятных событиях в Беларуси
Своевременное и точное информирование о нежелательных явлениях (НЯ) и серьезных нежелательных явлениях (СЯ) является краеугольным камнем эффективной системы фармаконадзора.
В Беларуси установлены специальные требования и процессы для обеспечения надлежащего документирования и передачи информации о безопасности фармацевтической продукции.
Требования к отчетности
Белорусские регуляторные органы утвердили стандартные формы для сообщения о подозрительных АЭ и неожиданных САЭ.
Эти формы предназначены для сбора основной информации, включая:
- Сведения о пациенте: инициалы, номер медицинской карты, возраст, пол, вес и другая необходимая демографическая информация.
- Информация о репортере: Полное имя, контактная информация и место работы медицинского работника или потребителя, сообщившего о происшествии.
- Сведения о продукте: название подозреваемого продукта, номер партии/лота, способ применения, показания и дозировка.
- Описание события: Подробный отчет о неблагоприятном событии, включая дату начала, продолжительность, степень тяжести и любую соответствующую историю болезни или сопутствующие препараты.
Для компаний крайне важно обеспечить точный учет и представление всех требуемых элементов данных, чтобы соответствовать местным нормативным требованиям.
Сроки представления отчетности в Беларуси
Сроки представления информации об АЭ и САЭ в Беларуси следующие:
- Серьезные нежелательные явления (SAEs): Компании должны подавать ускоренные отчеты о неожиданных SAE в течение 15 календарных дней после того, как им стало известно о событии.
- Несерьезные нежелательные явления: Отчеты о несерьезных АЭ должны быть включены в периодические отчеты об обновлении данных о безопасности (PSUR).
- Периодические отчеты об обновлении безопасности (PSURs): PSUR должны быть представлены в соответствии с установленными сроками, которые могут варьироваться в зависимости от продукта и его классификации риска. CETH содержит конкретные указания по срокам подачи PSUR в процессе регистрации продукта.
Компании должны иметь надежные процессы, обеспечивающие своевременное выявление, оценку и отчетность по АЭ и САЭ для соблюдения этих сроков.
Защита данных и конфиденциальность в Беларуси
GVP ЕАЭС и национальное законодательство Беларуси подчеркивают важность защиты персональных данных и сохранения конфиденциальности при работе с информацией о фармаконадзоре.
Компании, работающие в Беларуси, должны выполнять следующие меры:
- Защита персональных данных: Обеспечить, чтобы все персональные данные, собранные в процессе отчетности по АЭ, обрабатывались в соответствии с нормами защиты данных, включая получение необходимых согласий и применение соответствующих мер безопасности.
- Меры по обеспечению конфиденциальности: Установите протоколы для поддержания конфиденциальности сообщаемой информации во время передачи и хранения данных, такие как защищенные электронные системы, зашифрованные каналы связи и ограниченный доступ к конфиденциальным данным.
Несоблюдение требований по защите данных и конфиденциальности может привести к значительным штрафам и юридическим последствиям.

Каналы отчетности для отчетов об АЭ и САЭ в Беларуси
Все отчеты об АЭ и САЭ в Беларуси должны подаваться в ЦЭКТ, уполномоченный регуляторный орган, ответственный за прием и обработку данных фармаконадзора. В настоящее время отчеты могут быть представлены по следующим каналам:
- Бумажная отчетность: Компании могут отправить заполненные формы отчетности AE и SAE по почте или лично в офис CETH.
- Электронная отчетность: Хотя эта система еще не полностью внедрена, Беларусь изучает возможность создания системы электронной отчетности для упрощения процесса представления и обработки данных о фармаконадзоре.
По мере внедрения возможностей электронной отчетности компании должны быть готовы адаптировать свои процессы и системы для соответствия любым новым требованиям или рекомендациям, выпущенным ЦЭКТ.
Контрольный список отчетности для Беларуси
[ ] Используйте утвержденные формы отчетности по АЭ и САЭ
[ ] Точно фиксировать все необходимые элементы данных
[ ] Соблюдайте ускоренные сроки отчетности по SAE (15 календарных дней)
[ ] Включать несерьезные АЭ в периодические отчеты об обновлении безопасности (PSUR)
[ ] Принимать меры по защите персональных данных и сохранению конфиденциальности
[ ] Предоставлять отчеты в CETH по утвержденным каналам (в настоящее время в бумажном виде или лично)
[ ] Следить за обновлением системы электронной отчетности
Следуя этим требованиям к отчетности и лучшим практикам, международные компании могут обеспечить соблюдение обязательств по отчетности о неблагоприятных событиях в Беларуси и внести свой вклад в достижение общей цели обеспечения безопасности пациентов.
Сравнение практики применения GVP в Беларуси и ЕС
Хотя принятая Беларусью ГВП ЕАЭС в значительной степени гармонизирована с руководящими принципами Европейского союза (ЕС), существует ряд заметных различий в реализации и специфических требованиях к системе фармаконадзора.
Области гармонизации с GVP ЕС:
- Принятие схожих принципов и процессов для отчетности по АЭ, обнаружения сигналов и управления рисками
- Упор на защиту данных и меры по обеспечению конфиденциальности
- Требование к генеральному файлу системы фармаконадзора (PSMF)
Ключевые различия:
- Отсутствие единой базы данных ЕАЭС по ПВГ: В отличие от системы ЕС EudraVigilance , в ЕАЭС нет централизованной базы данных по фармаконадзору. Отчеты, поступающие из одной страны ЕАЭС, должны подаваться в другие страны-члены по отдельности.
- Потенциальные различия в сроках предоставления PSUR: В то время как в ЕС установлены сроки представления PSUR на основе классификаций риска продукции, в Беларуси могут быть установлены иные сроки для конкретных продуктов в процессе национальной регистрации.
- Местные формы и требования: В Беларуси существуют собственные формы, утвержденные на местном уровне, и особые требования к данным для отчетности по AE и SAE, которые могут незначительно отличаться от форм ЕС.
- Национальный и централизованный процесс выдачи разрешений на маркетинг: В отличие от централизованной процедуры получения разрешения на продажу в ЕС, в Беларуси применяется национальный процесс регистрации лекарственных средств, что может повлиять на требования и сроки фармаконадзора.
Таблица 2: Сравнение практик фармаконадзора ЕАЭС и ЕС
| Аспект | ЕАЭС (+ Беларусь) | Европейский союз |
| Руководящие принципы | ГВП ЕАЭС | ГВП ЕС |
| Централизованная база данных | Нет единой базы данных | EudraVigilance |
| Сроки проведения PSUR | Потенциальные различия | Гармонизированы в зависимости от степени риска продукта |
| Формы отчетности | Местные формы | Формы ЕС |
| Разрешение на маркетинг | Национальный процесс | Централизованная процедура |
Инспекции и аудиты фармацевтических компаний в Беларуси
Для обеспечения соблюдения правил фармаконадзора СЕТГ проводит регулярные инспекции и аудиты фармацевтических компаний, работающих в Беларуси.
Регуляторные инспекции СЭТГ:
- CETH имеет право проводить инспекции на местах для оценки системы и процессов фармаконадзора в компании.
- Проверки могут быть плановыми или проводиться в связи с особыми событиями, такими как проблемы безопасности продукции или несоответствия требованиям.
- Объем проверок может охватывать различные аспекты, включая процедуры отчетности по АЭ, ведение PSMF, деятельность по управлению рисками и целостность данных.
Подготовка к проверкам PVG:
- Компании должны поддерживать готовность к проверкам, регулярно пересматривая и обновляя документацию по фармаконадзору, процессы и программы обучения.
- Проведение внутренних аудитов и имитационных проверок поможет выявить потенциальные недостатки и области, требующие улучшения, до начала инспекции.
- Выделение специальной группы или персонала, ответственного за координацию и проведение проверок, может упростить этот процесс.
Роль аудитов в обеспечении соответствия GVP:
- В дополнение к проверкам регуляторных органов компаниям следует внедрить эффективную программу внутреннего аудита для постоянного мониторинга и оценки эффективности системы фармаконадзора.
- Аудиты позволяют выявить области несоответствия, неэффективность процессов и возможности для улучшения, что позволяет компаниям заблаговременно принимать корректирующие и предупреждающие меры.
- Регулярные аудиты демонстрируют приверженность компании к качеству и соблюдению требований, что может быть полезно во время проверок регулирующих органов.

Проблемы и лучшие практики фармаконадзора в Беларуси
Работа на белорусском рынке ставит перед международными фармацевтическими компаниями уникальные задачи, требующие стратегического подхода и следования лучшим практикам.
Языковые барьеры и культурные различия:
- Компании должны быть готовы к преодолению языковых барьеров и культурных различий при взаимодействии с местными регулирующими органами и медицинскими работниками.
- Использование местных ресурсов или сотрудничество с опытными консультантами по вопросам регулирования может помочь устранить эти пробелы и обеспечить эффективную коммуникацию.
Обеспечение последовательной глобальной отчетности:
- Поддержание последовательной практики отчетности на различных рынках имеет решающее значение для эффективного фармаконадзора и соблюдения нормативных требований.
- Компании должны создать стандартизированные глобальные процессы и системы для отчетности по АЭ, обнаружения сигналов и управления рисками, учитывая при этом местные требования.
Поддержание готовности к проверкам:
- Проверки регулирующих органов могут проводиться с минимальным уведомлением, поэтому компаниям необходимо постоянно поддерживать состояние готовности к проверке.
- Регулярное обучение, анализ процессов и имитация проверок помогут выявить и устранить потенциальные пробелы или области несоответствия.
Использование технологий и автоматизации:
- Внедрение надежного программного обеспечения для фармаконадзора и автоматизированных систем может оптимизировать процессы, повысить точность данных и обеспечить соблюдение сроков отчетности.
- По мере того как Беларусь будет развивать возможности электронной отчетности, компании должны быть готовы адаптировать свои системы и процессы соответствующим образом.
Важность изучения местных нормативных актов:
- Быть в курсе последних изменений в нормативно-правовой базе, руководящих документов и лучших отраслевых практик в Беларуси очень важно для обеспечения соответствия требованиям.
- Общение с местными экспертами в области регулирования, участие в отраслевых мероприятиях и мониторинг официальных сообщений могут дать ценные сведения и информацию.
Блок-схема: Процесс отчетности по АЭ в Беларуси
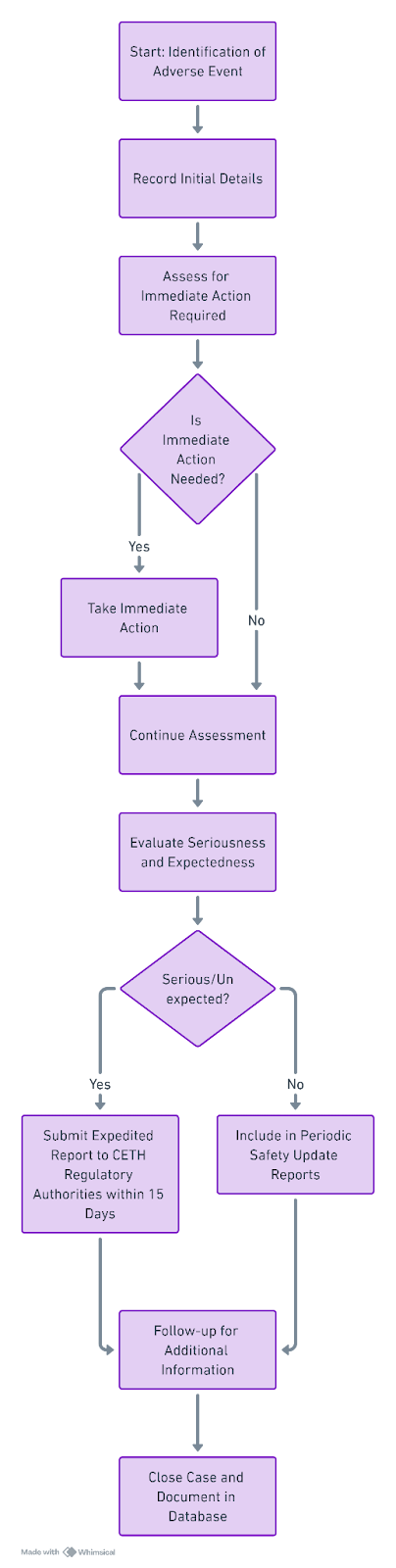
Ключевые термины и определения ГВП:
- Неблагоприятное событие (НС) : Любое нежелательное медицинское явление у пациента, принимающего фармацевтический препарат, которое не обязательно имеет причинно-следственную связь с лечением.
- Серьезное неблагоприятное событие (SAE): Нежелательное явление, которое приводит к смерти, угрожает жизни, требует госпитализации или продления существующей госпитализации, приводит к стойкой или значительной инвалидности/потере трудоспособности или является врожденной аномалией/врожденным дефектом.
- Периодический отчет о безопасности (PSUR): Периодический отчет, в котором обобщается мировой опыт безопасности фармацевтического продукта, включая все зарегистрированные АЭ и оценку риска и пользы.
- Главный файл системы фармаконадзора (PSMF): Подробный документ, описывающий систему фармаконадзора и ее соответствие требованиям GVP.
У вас есть вопросы о системе PVG в Беларуси?
Международные фармацевтические компании могут найти местный опыт неоценимым при навигации по белорусским процедурам регулирования и фармаконадзора. Специализированные агентства, такие как Delta Medical, имеющие офисы по всему региону Евразии, включая Беларусь, Азербайджан и Армению, Украину, Молдову и Центральную Азию, имеют все возможности, чтобы провести компании через тонкости нормативных требований.
Наше присутствие в Минске, связи с органами здравоохранения и опыт подачи региональных регуляторных документов делают работу на этих развивающихся рынках более удобной, чтобы компании могли сосредоточить основные усилия на разработке и коммерциализации лекарств. Наши целенаправленные рекомендации, дополняющие ваши внутренние возможности, являются ключом к эффективному выполнению требований Беларуси и обеспечению безопасности пациентов.
Будучи швейцарской фармацевтической компанией, Delta Medical может также заниматься маркетингом, логистикой, дистрибуцией и продажами, чтобы помочь компаниям достичь успешной коммерциализации на этих рынках.
Часто задаваемые вопросы о фармаконадзоре в Беларуси
Каковы основные законы и руководства по фармаконадзору в Беларуси?
- Закон о лекарственных средствах.
- Инструкция о порядке информирования о нежелательных реакциях.
- Инструкция по организации системы фармаконадзора.
- Принятие надлежащей практики фармаконадзора (НПП) Евразийского экономического союза (ЕАЭС).
Какова роль Центра экспертиз и испытаний в здравоохранении (ЦЭИТЗ)?
Каковы требования и формы отчетности о нежелательных явлениях в Беларуси?
Каковы сроки сообщения о нежелательных явлениях в Беларуси?
- Серьезные АЭ: 15 календарных дней.
- Несерьезные АЭ: Включены в периодические отчеты о безопасности (PSURs).
- PSURs: Сроки зависят от классификации риска продукта.