З населенням близько 10 мільйонів і зростаючим сектором охорони здоров’я в Азербайджані спостерігається постійне збільшення попиту на високоякісні лікарські засоби та медичні вироби.

Реєстрація лікарських засобів може бути складним завданням для міжнародних менеджерів з регуляторних питань, які не знайомі зі специфічними вимогами та процедурами країни. Для успішної реєстрації та виведення на ринок фармацевтичної продукції в Азербайджані вкрай важливо мати глибоке розуміння регуляторної бази, ключових органів влади та поетапного процесу реєстрації.
Ми сподіваємося надати міжнародним менеджерам з регуляторних питань необхідну інформацію та знання, які допоможуть їм впевнено вийти на азербайджанський ринок та оптимізувати процес реєстрації продукції.
Наприкінці цього посібника читачі матимуть чітке уявлення про:
- Основні регуляторні органи та їх роль у реєстрації лікарських засобів
- Законодавча база, що регулює обіг лікарських засобів в Азербайджані
- Види фармацевтичної продукції, що підлягають реєстрації
- Стандартна та прискорена процедури реєстрації та вимоги до них
- Післяреєстраційні зобов’язання та вимоги до фармаконагляду
- Маркування, пакування та правила імпорту/експорту
- Рекомендації для успішного виходу на ринок та дотримання вимог
Регуляторні органи та законодавча база в Азербайджані
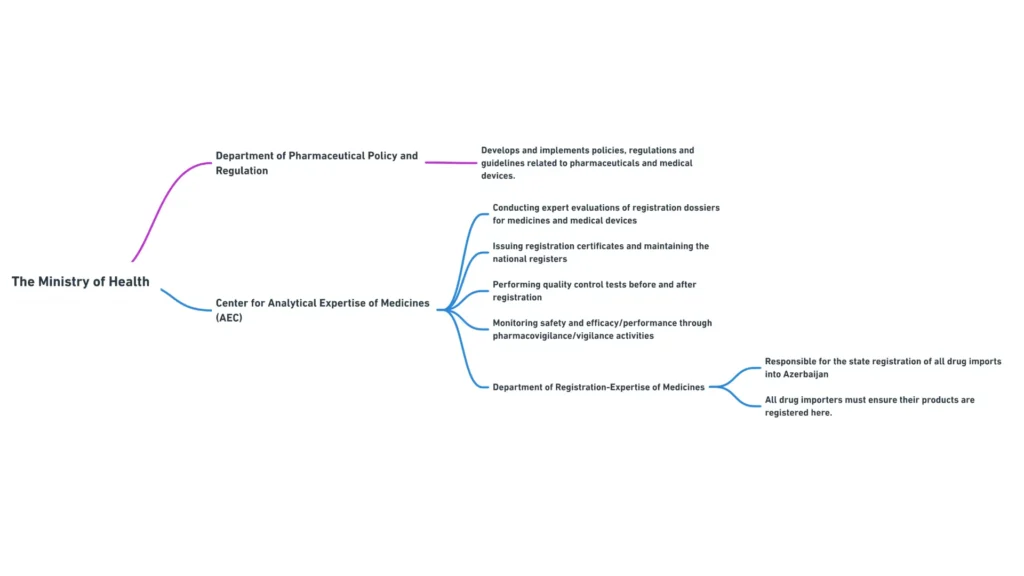
Міністерство охорони здоров’я Азербайджанської Республіки
Міністерство охорони здоров’я Азербайджанської Республіки є основним регуляторним органом, відповідальним за нагляд за сектором охорони здоров’я, включаючи регулювання фармацевтичної продукції. Основні обов’язки Міністерства включають
- Розробка та реалізація національної політики та стратегій, пов’язаних з охороною здоров’я та фармацевтикою
- Видача ліцензій на фармацевтичну діяльність, таку як виробництво, імпорт та оптова торгівля
- нагляд за реєстрацією, контролем якості та моніторингом безпеки лікарських засобів
- Координація з іншими державними органами та міжнародними організаціями з питань охорони здоров’я.
Міністерство охорони здоров’я складається з декількох департаментів та підпорядкованих органів, які відіграють певну роль у фармацевтичному регулюванні. Найбільш важливими для менеджерів з міжнародних регуляторних питань є наступні департаменти:
- Департамент фармацевтичної політики та регулювання: Відповідає за розробку та впровадження фармацевтичної політики, нормативно-правових актів та настанов
- Департамент ліцензування та сертифікації: займається ліцензуванням фармацевтичної діяльності та сертифікацією фармацевтичних фахівців
- Департамент міжнародних зв’язків: Координує співпрацю Міністерства з міжнародними організаціями та іноземними регуляторними органами
Міністерство охорони здоров’я Азербайджану та відомства, що працюють з фармацевтичними препаратами
Центр аналітичної експертизи лікарських засобів
Центр аналітичної експертизи лікарських засобів є підпорядкованим Міністерству охорони здоров’я органом, який відіграє вирішальну роль у реєстрації та контролі якості фармацевтичної продукції в Азербайджані. Основні функції Центру включають
- проведення експертизи реєстраційних досьє на лікарські засоби
- Видача реєстраційних посвідчень та ведення національного реєстру лікарських засобів
- Проведення випробувань з контролю якості лікарських засобів до та після реєстрації
- Моніторинг безпеки та ефективності зареєстрованих лікарських засобів шляхом здійснення фармаконагляду.
Центр аналітичної експертизи лікарських засобів є основною контактною особою для менеджерів з міжнародних регуляторних питань під час процесу реєстрації. Для забезпечення безперебійного процесу реєстрації важливо підтримувати чіткий зв’язок з Центром та оперативно реагувати на будь-які запити або прохання про надання додаткової інформації.
Відповідне законодавство та нормативні акти
Нормативно-правова база, що регулює фармацевтичну діяльність в Азербайджані, складається із законів, указів та підзаконних актів, які встановлюють вимоги та процедури щодо реєстрації, виробництва, імпорту та дистрибуції лікарських засобів.
До найбільш важливих законодавчих актів, з якими слід ознайомитися менеджерам з міжнародних регуляторних питань, відносяться наступні:
- Закон “Про лікарські засоби” (2006): основний закон, що регулює фармацевтичну продукцію в Азербайджані, який охоплює такі аспекти, як реєстрація, виробництво, контроль якості та фармаконагляд. З моменту прийняття закону до нього було внесено кілька поправок з метою приведення його у відповідність до міжнародних стандартів та найкращих практик.
- Постанова № 108 “Про правила державної реєстрації лікарських засобів” (2007)<40>: Викладає детальні вимоги та процедури для реєстрації фармацевтичної продукції, включаючи зміст реєстраційного досьє, строки та збори.
- Постанова № 137 “Про правила проведення експертизи лікарських засобів” (2007): Визначає процедури та критерії експертної оцінки реєстраційних досьє Центром аналітичної експертизи лікарських засобів.
- Постанова № 137 “Про правила імпорту та експорту лікарських засобів” (2007)<48>: встановлює вимоги та процедури імпорту та експорту фармацевтичної продукції, включаючи ліцензування, митне оформлення та документацію.
Окрім цих ключових законодавчих актів, менеджери з міжнародних регуляторних питань також повинні бути в курсі будь-яких змін, додаткових нормативно-правових актів або методичних документів, що видаються Міністерством охорони здоров’я або Центром аналітичної експертизи лікарських засобів.
Регулярне відвідування офіційних веб-сайтів та інформаційних бюлетенів цих органів допоможе забезпечити відповідність найсучаснішим вимогам.
Види фармацевтичної продукції, що підлягають реєстрації
В Азербайджані більшість фармацевтичних продуктів повинні бути зареєстровані в Центрі аналітичної експертизи лікарських засобів (ЦАЕЛ), перш ніж вони можуть бути випущені на ринок і продані. Вимога реєстрації поширюється як на імпортовані, так і на вітчизняні лікарські засоби.
Основні види фармацевтичної продукції, що підлягають реєстрації, включають
- Оригінальні лікарські засоби: Інноваційні лікарські засоби, що містять нові діючі речовини або нові комбінації діючих речовин, які раніше не були зареєстровані в Азербайджані.
- Генеричні лікарські засоби: Фармацевтична продукція, яка містить ті ж самі активні речовини, лікарську форму та силу дії, що і вже зареєстрований оригінальний лікарський засіб, і призначена для взаємозамінності з оригінальним продуктом.
- Комбіновані препарати: Лікарські засоби, які містять дві або більше діючих речовин у фіксованій дозі, де кожна діюча речовина робить свій внесок у загальний терапевтичний ефект.
- Лікарські форми та дозування: Різні лікарські форми (наприклад, таблетки, капсули, сиропи) та дозування однієї і тієї ж активної речовини вимагають окремої реєстрації, навіть якщо вони виробляються одним виробником.
Однак, певні види лікарських засобів можуть бути звільнені від вимоги реєстрації, наприклад, лікарські засоби, що виготовляються в аптеках:
- лікарські засоби, що виготовляються в аптеках за рецептом лікаря для індивідуальних пацієнтів
- Ліки, що ввозяться для особистого користування в кількості, що не перевищує встановленого ліміту
- Лікарські засоби, що ввозяться з метою проведення клінічних випробувань або наукових досліджень, за попереднім погодженням з Міністерством охорони здоров’я.
Менеджерам з міжнародних регуляторних питань необхідно ретельно оцінити свій портфель продуктів і визначити, які з них потребують реєстрації в Азербайджані. Ця оцінка повинна враховувати такі фактори, як склад, призначення та потенційний ринковий попит для кожного продукту.
| Тип продукту | Вимоги до реєстрації |
| Оригінальні лікарські засоби | Так |
| Генеричні ліки | Так |
| Комбіновані продукти | Так |
| Різні лікарські форми та дозування | Потрібна окрема реєстрація |
| Ліки, що виготовляються в аптеках для індивідуальних пацієнтів | Звільняються |
| Ліки, що ввозяться для особистого користування (в обмеженій кількості) | Звільняються |
| Ліки, що ввозяться для клінічних випробувань або досліджень (за наявності дозволу) | Звільняються |
Процедури та строки реєстрації лікарських засобів в Азербайджані
Для того, щоб зареєструвати лікарський засіб в Азербайджані, менеджери з міжнародних регуляторних питань повинні дотримуватися відповідної процедури реєстрації та подати повне досьє до Центру аналітичної експертизи лікарських засобів.
Процес реєстрації складається з двох основних процедур: стандартної процедури реєстрації та прискореної процедури реєстрації.
Стандартна процедура реєстрації лікарських засобів в Азербайджані
Стандартна процедура реєстрації – це найпоширеніший шлях для реєстрації фармацевтичної продукції в Азербайджані. Ця процедура передбачає ретельну оцінку якості, безпеки та ефективності продукту Центром аналітичної експертизи лікарських засобів.
Основні етапи та вимоги стандартної процедури реєстрації є наступними:
- Подання досьє: Заявник повинен подати повне реєстраційне досьє до Центру аналітичної експертизи лікарських засобів. Досьє повинно бути підготовлене відповідно до формату Загального технічного документа (CTD) і включати наступні модулі:
- Адміністративна інформація (Модуль 1)
- Дані про якість (Модуль 2)
- Неклінічні дані (Модуль 3)
- Клінічні дані (Модуль 4)
- План фармаконагляду та управління ризиками (Модуль 5)
- Досьє подається в паперовому та електронному вигляді разом з необхідним реєстраційним збором.
- Попередня експертиза: Після отримання досьє Центр аналітичної експертизи лікарських засобів проводить попередню експертизу, щоб переконатися, що заявка є повною і відповідає формальним вимогам. Якщо будуть виявлені будь-які недоліки, заявник буде повідомлений про це і йому буде надано певний термін для їх усунення.
- Спеціалізована експертиза: Після того, як досьє проходить попередній розгляд, воно проходить спеціалізовану експертизу, яка передбачає детальну оцінку даних щодо якості, безпеки та ефективності продукту групою експертів. Цей етап може включати
- Оцінку фармацевтичної та аналітичної документації
- Аналіз звітів про неклінічні та клінічні дослідження
- Оцінка профілю “користь-ризик” продукту
- Інспекція виробничої дільниці (дільниць) на відповідність вимогам належної виробничої практики (GMP)
- На цьому етапі Центр аналітичної експертизи лікарських засобів може запросити у заявника додаткову інформацію або роз’яснення. Щоб уникнути затримок у процесі реєстрації, дуже важливо оперативно та ретельно відповідати на ці запити.
- Рішення та видача свідоцтва: За результатами спеціалізованої експертизи Центр аналітичної експертизи лікарських засобів приймає рішення про схвалення або відхилення заявки на реєстрацію. У разі позитивного рішення заявник отримає реєстраційне посвідчення, дійсне протягом п’яти років. Зареєстрований продукт також буде внесено до національного реєстру лікарських засобів.
Стандартна процедура реєстрації зазвичай займає від 6 до 12 місяців від дати подання досьє до видачі реєстраційного посвідчення.
Однак цей термін може змінюватися залежно від складності продукту, якості поданого досьє та завантаженості Центру аналітичної експертизи лікарських засобів.
Прискорена процедура реєстрації в Азербайджані
У липні 2020 року Азербайджан запровадив прискорену процедуру реєстрації для спрощення процесу реєстрації певних категорій фармацевтичної продукції. Ця процедура має на меті скоротити терміни реєстрації, забезпечуючи при цьому якість, безпеку та ефективність продукції.
Щоб отримати право на прискорену процедуру реєстрації, лікарський засіб повинен відповідати одному з наступних критеріїв:
- Продукт вже зареєстрований в країнах із суворими регуляторними органами (наприклад, ЄС, США, Японія, Австралія, Канада)
- Прекваліфікований Всесвітньою організацією охорони здоров’я (ВООЗ)
- Продукт призначений для лікування рідкісних захворювань або станів з високими незадоволеними медичними потребами
За прискореною процедурою реєстрації Центр аналітичної експертизи лікарських засобів визначатиме пріоритетність оцінки відповідних продуктів і прагнутиме завершити процес реєстрації протягом 3-6 місяців.
Однак, заявник все одно повинен подати повне реєстраційне досьє і оперативно відповідати на будь-які запити або прохання про надання додаткової інформації від органів влади.
Важливо зазначити, що прискорена процедура реєстрації не звільняє продукти від вимоги продемонструвати якість, безпеку та ефективність. Застосовуються ті ж науково-технічні стандарти, що і в стандартній процедурі реєстрації.
Відмова в реєстрації лікарського засобу в Азербайджані
У деяких випадках Центр аналітичної експертизи лікарських засобів може відмовити в реєстрації лікарського засобу. Найпоширенішими підставами для відмови є
- Недостатні або неадекватні дані, що демонструють якість, безпеку або ефективність продукту
- невідповідність вимогам належної виробничої практики (GMP) або іншим відповідним стандартам
- Спотворення або фальсифікація інформації в реєстраційному досьє
- Неусунення недоліків або ненадання додаткової інформації на вимогу органів влади у встановлений термін.
Якщо заявка на реєстрацію відхилена, заявник отримає письмове повідомлення із зазначенням причин відмови. Заявник може оскаржити рішення протягом 30 днів шляхом подання письмового запиту до Міністерства охорони здоров’я.
Апеляція буде розглянута експертною комісією вищого рівня, яка прийме остаточне рішення щодо реєстраційної заяви.
Зміни до зареєстрованих лікарських засобів
Після того, як лікарський засіб було зареєстровано в Азербайджані, про будь-які зміни у складі, виробничому процесі, маркуванні або інших аспектах необхідно повідомляти до Центру аналітичної експертизи лікарських засобів.
Тип зміни визначає, чи потрібно подавати повідомлення або перереєстрацію.
Типи змін, що потребують повідомлення або перереєстрації
- Зміни у складі та процесі виробництва:
- Незначні зміни у складі або процесі виробництва, які не впливають на якість, безпеку або ефективність лікарського засобу, можуть вимагати лише повідомлення компетентних органів.
- Значні зміни, такі як додавання або вилучення діючої речовини, суттєві зміни у виробничому процесі або зміна місця виробництва, можуть вимагати перереєстрації.
- Зміна маркування та упаковки:
- Незначні зміни в маркуванні або упаковці, такі як оновлення контактної інформації виробника або додавання штрих-коду, можуть вимагати лише повідомлення.
- Значні зміни, такі як зміна назви, показань, дозування або умов зберігання, можуть вимагати перереєстрації.
- Показання та протипоказання:
- Будь-які зміни в затверджених показаннях, протипоказаннях або інших аспектах інструкції для медичного застосування лікарського засобу потребують перереєстрації.
Процедура подання запитів на внесення змін до реєстраційних матеріалів в Азербайджані
Щоб повідомити Центр аналітичної експертизи лікарських засобів про зміну або подати запит на перереєстрацію, власник реєстраційного посвідчення повинен подати письмову заяву разом із супровідною документацією.
Заява повинна чітко описувати характер та обсяг змін, а також містити докази того, що ці зміни не впливають негативно на якість, безпеку або ефективність лікарського засобу.
Центр аналітичної експертизи лікарських засобів розгляне заяву на внесення змін і прийме рішення про те, чи затвердити зміну, чи вимагати додаткову інформацію. Терміни розгляду залежать від складності зміни та завантаженості органів влади.
| Тип змін | Повідомлення | Перереєстрація |
| Незначні зміни у складі або виробництві | ✓ | |
| Значні зміни у складі або виробництві | ✓ | |
| Незначні зміни маркування або упаковки | ✓ | |
| Значні зміни в маркуванні або пакуванні | ✓ | |
| Зміна показань або протипоказань | ✓ |
Невиконання вимог щодо повідомлення про зміни або перереєстрації може призвести до штрафних санкцій, призупинення або скасування реєстрації продукту.
Поновлення та анулювання реєстрації лікарських засобів
В Азербайджані реєстрація лікарського засобу дійсна протягом п’яти років з дати видачі реєстраційного посвідчення. Щоб продовжити реалізацію лікарського засобу після закінчення терміну дії реєстрації, власник реєстраційного посвідчення повинен подати заявку на його перереєстрацію.
Процес подання заяви на перереєстрацію та вимоги до неї в Азербайджані
Заявка на перереєстрацію повинна бути подана до Центру аналітичної експертизи лікарських засобів щонайменше за 90 днів до закінчення терміну дії поточної реєстрації.
Заява повинна містити наступні документи:
- Заповнену форму заявки на поновлення
- Копія чинного реєстраційного посвідчення
- Оновлені дані з якості, безпеки та ефективності, включаючи:
- Періодичні звіти з безпеки (PSUR)
- Дані постмаркетингового нагляду
- Будь-які нові клінічні або неклінічні дослідження, проведені з моменту первинної реєстрації
- Декларація, яка підтверджує, що склад, виробничий процес та інші аспекти продукту залишаються незмінними, або детально описує будь-які зміни, внесені з моменту первинної реєстрації.
- Зразки лікарського засобу та референтні стандарти для проведення контролю якості, якщо цього вимагають органи влади.
Центр аналітичної експертизи лікарських засобів розгляне заяву про перереєстрацію та прийме рішення про надання дозволу на перереєстрацію протягом 60 днів. Якщо органам влади знадобиться додаткова інформація або роз’яснення, строк розгляду може бути подовжено.
Підстави для скасування реєстрації лікарського засобу
Центр аналітичної експертизи лікарських засобів може скасувати реєстрацію лікарського засобу за таких обставин:
- Власник реєстраційного посвідчення не подав заяву на перереєстрацію до закінчення терміну дії поточної реєстрації
- Продукт визнано небезпечним, неефективним або неякісним на підставі даних постмаркетингового нагляду або інших доказів
- Власник реєстраційного посвідчення не виконує зобов’язання з фармаконагляду або інші післяреєстраційні вимоги
- Препарат не продається в Азербайджані протягом трьох років поспіль без поважної причини
- Власник реєстраційного посвідчення просить скасувати реєстрацію
Якщо Центр аналітичної експертизи лікарських засобів має намір скасувати реєстрацію лікарського засобу, він повідомляє про це власника реєстраційного посвідчення в письмовій формі з викладенням причин запропонованого скасування.
Власник реєстраційного посвідчення має право оскаржити рішення протягом 30 днів шляхом подання письмової заяви до Міністерства охорони здоров’я.
Наслідки непродовження або анулювання
Якщо реєстрація лікарського засобу не поновлюється або анулюється, власник реєстраційного посвідчення зобов’язаний
- Припинити будь-яку діяльність з маркетингу та дистрибуції лікарського засобу в Азербайджані
- Вилучити продукт з ринку в терміни, визначені уповноваженими органами
- повідомити медичних працівників та інші зацікавлені сторони про відмову від поновлення або відкликання
- Співпрацювати з органами влади щодо будь-якого відкликання або інших заходів зі зниження ризику, які вважаються необхідними для захисту здоров’я населення.
Невиконання цих зобов’язань може призвести до санкцій, включаючи штрафи та судові позови.
Фармаконагляд та постмаркетингові зобов’язання
Для забезпечення постійної безпеки та ефективності зареєстрованих лікарських засобів власники реєстраційних посвідчень в Азербайджані повинні дотримуватися зобов’язань з фармаконагляду та інших постмаркетингових зобов’язань.
Вимоги до системи фармаконагляду в Азербайджані
Власники реєстраційних посвідчень повинні створити та підтримувати систему фармаконагляду, яка відповідає вимогам, викладеним у Законі “Про лікарські засоби” та відповідних нормативно-правових актах.
Ключові елементи системи фармаконагляду включають
- Призначення місцевої контактної особи: Власник реєстраційного посвідчення повинен призначити кваліфіковану особу, відповідальну за здійснення фармаконагляду (УОВФ), яка проживає в Азербайджані і є основною контактною особою для органів влади з питань фармаконагляду.
- Повідомлення про побічні реакції: Власник реєстраційного посвідчення повинен повідомляти про всі серйозні побічні реакції, пов’язані із застосуванням його продукції в Азербайджані, до Центру аналітичної експертизи лікарських засобів протягом 15 днів з моменту отримання інформації. Про несерйозні побічні явища необхідно повідомляти протягом 90 днів.
- Виявлення сигналів та управління ризиками: Власник реєстраційного посвідчення повинен постійно контролювати профіль безпеки своєї продукції та виявляти будь-які нові ризики або зміни у співвідношенні користь/ризик. У разі виявлення нового сигналу з безпеки власник реєстраційного посвідчення повинен повідомити про це компетентні органи та вжити відповідних заходів з управління ризиками.
Періодичні звіти з безпеки (ПЗБ)
Власники реєстраційних посвідчень повинні подавати періодичні звіти з безпеки (PSUR) до Центру аналітичної експертизи лікарських засобів згідно з наступним графіком:
- Кожні 6 місяців протягом перших 2 років після реєстрації
- Щорічно протягом наступних 2 років
- Кожні 3 роки після цього
PSUR повинен містити всебічний огляд профілю безпеки лікарського засобу, включаючи аналіз усіх небажаних явищ, про які повідомлялося у світі, будь-яких нових виявлених сигналів з безпеки, а також будь-яких змін у співвідношенні користь/ризик.
Інспекції та аудити власників реєстраційних посвідчень
Центр аналітичної експертизи лікарських засобів може проводити інспекції та аудити власників реєстраційних посвідчень з метою забезпечення дотримання ними зобов’язань з фармаконагляду та інших постмаркетингових зобов’язань.
Під час інспекції або аудиту органи влади можуть перевіряти систему фармаконагляду власника реєстраційного посвідчення, процеси звітування про побічні реакції, плани управління ризиками та іншу відповідну документацію.
Власники реєстраційних посвідчень повинні повною мірою співпрацювати з органами влади під час інспекцій та аудитів і негайно усувати будь-які виявлені недоліки або невідповідності.
Штрафи за недотримання вимог
Невиконання зобов’язань з фармаконагляду та інших постмаркетингових зобов’язань може призвести, зокрема, до штрафних санкцій:
- Штрафи та адміністративні санкції
- Призупинення або скасування реєстрації продукту
- Кримінальне переслідування у випадках серйозних порушень або шахрайства
Щоб уникнути цих наслідків, менеджери з міжнародних регуляторних питань повинні переконатися, що їхні компанії мають надійні системи фармаконагляду, а також регулярно переглядати та оновлювати свої процеси для забезпечення відповідності азербайджанським вимогам.
Вимоги до маркування та пакування в Азербайджані
Фармацевтична продукція, що продається в Азербайджані, повинна відповідати певним вимогам до маркування та пакування, щоб забезпечити доступ медичних працівників і пацієнтів до точної та повної інформації про продукт.
Вимоги до мови та змісту
Всі етикетки та пакувальні матеріали для фармацевтичної продукції повинні бути азербайджанською мовою.
На додаток до назви продукту, назви та адреси виробника, на етикетці та вкладиші до упаковки повинна міститися наступна інформація:
- Склад, включаючи діючі та допоміжні речовини
- Лікарська форма та сила дії
- Показання та протипоказання
- Дозування та спосіб застосування
- Попередження та запобіжні заходи
- Побічні реакції
- Умови зберігання та термін придатності
- Номер серії та термін придатності
- Реєстраційний номер та дата реєстрації
Особливі вимоги до підконтрольних речовин та рецептурних лікарських засобів
Фармацевтична продукція, що містить підконтрольні речовини або відпускається за рецептом, повинна містити додаткові елементи маркування, такі як
- Міжнародна непатентована назва (МНН) підконтрольної речовини
- концентрація підконтрольної речовини
- Рецептурний статус продукту (наприклад, “Тільки за рецептом”)
- Спеціальні вимоги до зберігання та поводження з продуктом
Процес затвердження маркування та пакувальних матеріалів
Перш ніж фармацевтичний продукт може бути випущений на ринок Азербайджану, маркування та пакувальні матеріали повинні бути затверджені Центром аналітичної експертизи лікарських засобів (ЦАЕЛ). Власник реєстраційного посвідчення повинен подати зразки запропонованого маркування та упаковки разом з реєстраційним досьє або в рамках післяреєстраційного запиту на внесення змін.
Центр аналітичної експертизи лікарських засобів розгляне маркування та пакувальні матеріали, щоб переконатися, що вони відповідають вимогам щодо мови та змісту, а також точно відображають затверджену інформацію для медичного застосування лікарського засобу.
Якщо компетентні органи вимагають внесення змін до маркування або упаковки, власник реєстраційного посвідчення повинен переглянути матеріали та повторно подати їх на затвердження.
Правила імпорту та експорту
Менеджери з міжнародних регуляторних питань також повинні бути обізнані з правилами імпорту та експорту фармацевтичної продукції в Азербайджані.
Ліцензійні вимоги до імпортерів та експортерів фармацевтичної продукції
Компанії, що займаються імпортом або експортом фармацевтичної продукції в Азербайджані, повинні отримати відповідні ліцензії від Міністерства охорони здоров’я.
Основні ліцензійні вимоги включають
- Ліцензія імпортера: Компанії, які імпортують фармацевтичну продукцію в Азербайджан, повинні отримати ліцензію імпортера. Щоб мати право на отримання ліцензії імпортера, компанія повинна мати
- Зареєстровану юридичну особу в Азербайджані
- Відповідні приміщення та обладнання для зберігання та обробки фармацевтичної продукції
- Кваліфікований персонал, включаючи відповідального фармацевта
- Система контролю якості, що відповідає вимогам Належної практики дистрибуції (GDP)
- Ліцензія експортера: Компанії, які експортують фармацевтичну продукцію з Азербайджану, повинні отримати ліцензію експортера. Щоб мати право на отримання експортної ліцензії, компанія повинна мати
- Зареєстровану юридичну особу в Азербайджані
- Діюча ліцензія на виробництво або контракт з ліцензованим виробником
- Відповідні приміщення та обладнання для зберігання та поводження з фармацевтичною продукцією
- Кваліфікований персонал, включаючи відповідального фармацевта
- Система контролю якості, що відповідає вимогам Належної виробничої практики (GMP) та Належної практики дистрибуції (GDP)
Процедури митного оформлення та документація
При імпорті або експорті фармацевтичної продукції компанії повинні дотримуватися процедур митного оформлення та надавати необхідну документацію, зокрема
- Комерційний інвойс
- Пакувальний лист
- Сертифікат аналізу
- Сертифікат походження
- Свідоцтво про реєстрацію або дозвіл на імпорт, залежно від ситуації
- Інші документи, що вимагаються митними органами
Особливі вимоги до підконтрольних речовин та продуктів холодового ланцюга
Фармацевтична продукція, що містить підконтрольні речовини або потребує зберігання та транспортування в умовах холодового ланцюга, підлягає додатковим вимогам щодо імпорту та експорту. Для підконтрольних речовин компанії повинні отримувати дозвіл на імпорт або експорт від Міністерства охорони здоров’я на кожну партію.
Для продуктів холодового ланцюга компанії повинні забезпечити зберігання і транспортування продукції при дотриманні необхідних температурних умов і надати докази моніторингу температури під час митного оформлення.
Останні думки щодо реєстрації лікарських засобів в Азербайджані
Для успішного проходження фармацевтичних процедур в Азербайджані менеджери з міжнародних регуляторних питань повинні враховувати наступні рекомендації:
- Залучайте місцевих регуляторних експертів та партнерів: Співпраця з місцевими регуляторними експертами та партнерами, такими як Delta Medical, може допомогти міжнародним компаніям зрозуміти та виконати вимоги Азербайджану, уникнути поширених помилок та прискорити процес реєстрації.
- Плануйте заздалегідь і виділяйте достатньо часу для реєстрації: Процес реєстрації в Азербайджані може бути тривалим і складним, особливо для інноваційних продуктів. Щоб уникнути затримок з виходом на ринок, міжнародні компанії повинні планувати заздалегідь і виділити достатньо часу на підготовку досьє, подання та розгляд, щоб уникнути затримок з виходом на ринок.
- Надавати пріоритет продуктам з високим ринковим потенціалом: Щоб максимізувати рентабельність інвестицій, міжнародні компанії повинні надавати пріоритет реєстрації продуктів з високим ринковим потенціалом в Азербайджані, беручи до уваги такі фактори, як поширеність захворювань, незадоволені медичні потреби та конкуренція.
- Забезпечити дотримання післяреєстраційних вимог: Міжнародні компанії повинні створити надійні системи та процеси фармаконагляду для дотримання післяреєстраційних вимог в Азербайджані. Регулярний перегляд і оновлення цих систем може допомогти уникнути штрафних санкцій і забезпечити безперебійну роботу бізнесу.
- Будьте в курсі регуляторних змін та оновлень: Фармацевтичний регуляторний ландшафт Азербайджану постійно розвивається, періодично з’являються нові нормативні акти та інструкції. Менеджери з міжнародних регуляторних питань повинні бути в курсі цих змін і відповідно адаптувати свої стратегії та процеси.
Дотримуючись цих рекомендацій та тісно співпрацюючи з місцевими партнерами, міжнародні компанії можуть успішно орієнтуватися на фармацевтичному ринку Азербайджану та сприяти покращенню доступу пацієнтів до безпечних, ефективних та якісних лікарських засобів.


