Азербайджан, быстро развивающаяся страна в Кавказском регионе, представляет собой многообещающую возможность для международных компаний по производству медицинских изделий, стремящихся расширить свое присутствие на рынке. С населением около 10 миллионов человек и растущим сектором здравоохранения в Азербайджане наблюдается увеличение спроса на инновационные и высококачественные медицинские изделия.

Управление нормативно-правовой базой для медицинских изделий в Азербайджане может быть сложной задачей для международных менеджеров по регуляторным вопросам, которые не знакомы с особыми требованиями и процедурами страны.
Цель данного исчерпывающего руководства — предоставить международным менеджерам по регуляторным вопросам необходимую информацию и знания, чтобы уверенно работать на азербайджанском рынке и оптимизировать свои усилия по регистрации устройств.
Мы надеемся, что вы получите четкое представление о:
- Основные регулирующие органы и их роль в регистрации медицинских изделий
- Законодательная база, регулирующая медицинские изделия в Азербайджане
- Система классификации медицинских изделий и ее последствия для регистрации
- Общие и упрощенные процедуры регистрации и их требования
- Пострегистрационные обязательства, включая маркировку, бдительность и постмаркетинговое наблюдение
- Правила импорта и экспорта медицинских изделий
- Рекомендации по успешному выходу на рынок и соблюдению требований
Регулирующие органы и правовая база в Азербайджане
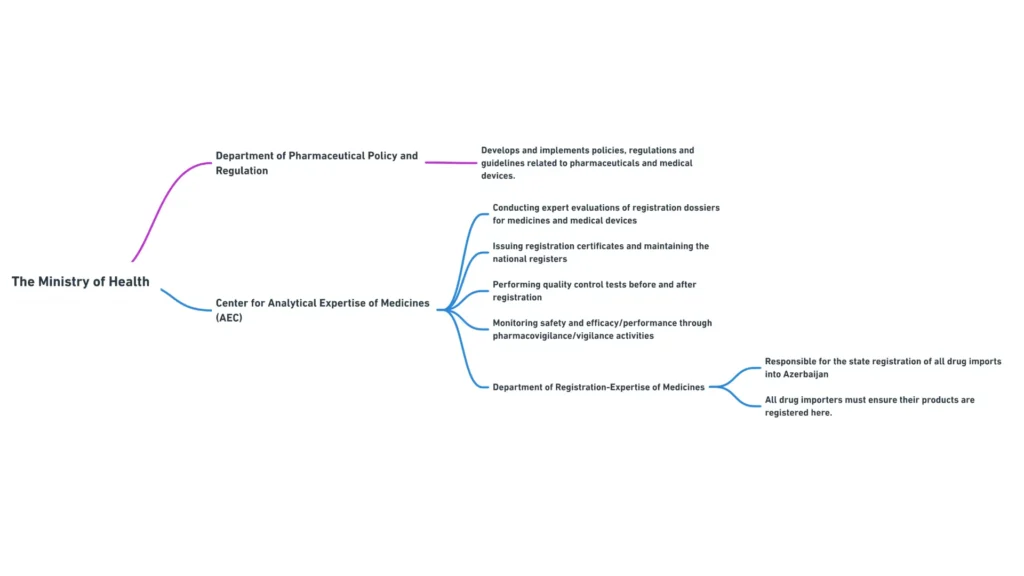
Министерство здравоохранения Азербайджанской Республики
Министерство здравоохранения Азербайджанской Республики является основным регулирующим органом, ответственным за надзор за сектором здравоохранения, включая регулирование медицинских изделий. Основные обязанности Министерства включают:
- Разработка и реализация национальной политики и стратегий, связанных со здравоохранением и медицинскими изделиями
- Выдача лицензий на деятельность, связанную с медицинскими изделиями, такую как производство, импорт и распространение
- Надзор за регистрацией, контролем качества и безопасностью медицинских изделий
- Сотрудничество с международными организациями и зарубежными регулирующими органами по вопросам, связанным с медицинскими изделиями.
Министерство здравоохранения включает в себя несколько департаментов и подчиненных им органов, которые играют определенную роль в регулировании медицинских изделий. Наиболее актуальными для менеджеров по международным регуляторным вопросам являются следующие департаменты:
- Департамент фармацевтической политики и регулирования: Отвечает за разработку и внедрение политики, правил и рекомендаций, связанных с медицинскими изделиями и фармацевтическими препаратами.
- Департамент лицензирования и сертификации: Занимается лицензированием деятельности, связанной с медицинскими изделиями, и сертификацией специалистов по медицинским изделиям.
- Департамент международных отношений: Координирует сотрудничество Министерства с международными организациями и зарубежными регулирующими органами в области медицинских изделий.
Структура Министерства здравоохранения Азербайджана и департаменты
Центр аналитической экспертизы лекарственных средств
Центр аналитической экспертизы лекарственных средств, подведомственный Министерству здравоохранения, играет важнейшую роль в регистрации и контроле качества медицинских изделий в Азербайджане.
К основным функциям Центра относятся:
- Проведение экспертизы регистрационных досье на медицинские изделия
- Выдача регистрационных удостоверений и ведение национального реестра медицинских изделий
- Проведение испытаний по контролю качества медицинских изделий до и после регистрации
- Мониторинг безопасности и эффективности зарегистрированных медицинских изделий с помощью мероприятий по бдительности.
Центр аналитической экспертизы лекарственных средств является основным контактным лицом для международных менеджеров по регуляторным вопросам в процессе регистрации.
Поддержание четкой связи с Центром и оперативное реагирование на любые запросы или просьбы о предоставлении дополнительной информации имеет решающее значение для обеспечения беспрепятственного процесса регистрации.
Соответствующее законодательство и нормативные акты
Законодательная база, регулирующая медицинские изделия в Азербайджане, состоит из законов, указов и постановлений, которые устанавливают требования и процедуры регистрации, производства, импорта и распространения медицинских изделий.
Наиболее важные законодательные акты, о которых должны знать международные менеджеры по регуляторным вопросам, включают:
- Закон о лекарственных средствах (2006 г.): Основной закон, регулирующий медицинские изделия и фармацевтическую продукцию в Азербайджане. Он охватывает такие аспекты, как регистрация, производство, контроль качества и бдительность. С момента принятия в закон было внесено несколько поправок для приведения его в соответствие с международными стандартами и передовым опытом.
- Указ № 108 о правилах государственной регистрации лекарственных средств (2007 г.): Определяет подробные требования и процедуры регистрации медицинских изделий и фармацевтической продукции, включая содержание регистрационных досье, сроки и сборы.
- Постановление № 137 о правилах проведения экспертизы лекарственных средств (2007 г.): Определяет процедуры и критерии экспертной оценки регистрационных досье Центром аналитической экспертизы лекарственных средств.
- Постановление № 137 «О правилах импорта и экспорта лекарственных средств» (2007): устанавливает требования и процедуры импорта и экспорта медицинских изделий и фармацевтической продукции, включая лицензирование, таможенное оформление и документацию.
Помимо этих ключевых законодательных актов, менеджеры по международным регуляторным вопросам также должны быть в курсе всех изменений, дополнительных правил или руководящих документов, выпущенных Министерством здравоохранения или Центром аналитической экспертизы лекарственных средств.
Регулярное обращение к официальным сайтам и информационным бюллетеням этих органов может помочь обеспечить соблюдение самых современных требований.
Классификация медицинских изделий в Азербайджане
В Азербайджане действует система классификации медицинских изделий по степени риска, которая в значительной степени гармонизирована с системой классификации Европейского союза в соответствии с Положением о медицинских изделиях (MDR). Классификация медицинского изделия определяет применимые процедуры оценки соответствия и уровень регуляторного надзора, необходимого для регистрации.
Правила и критерии классификации
Медицинские изделия делятся на четыре категории в зависимости от их предназначения, продолжительности использования, инвазивности и потенциального риска для пациента или пользователя.
Правила классификации учитывают такие факторы, как:
- Способ действия устройства и источник энергии
- Степень и продолжительность контакта с телом человека
- Возможность причинения вреда или травм в случае неисправности или неправильного использования
- Зависимость устройства от источника питания или программного обеспечения
- Использование устройства в сочетании с другими устройствами или веществами.
Существует четыре категории классификации:
- Класс I: Устройства с низким уровнем риска, которые не контактируют с пациентом или имеют лишь кратковременный контакт с неповрежденной кожей (например, нестерильные бинты, смотровые перчатки и приспособления для ходьбы).
- Класс IIa: Устройства с низким или средним уровнем риска, которые контактируют с поврежденной кожей, вводятся в отверстия тела или используются для кратковременных инвазивных процедур (например, отсасывающее оборудование, слуховые аппараты и стоматологические пломбировочные материалы).
- Класс IIb: Устройства среднего и высокого риска, используемые для длительных инвазивных процедур, имплантируемые или способные нанести травму в случае неисправности (например, аппараты искусственной вентиляции легких, ортопедические имплантаты и инфузионные насосы).
- Класс III: Устройства высокого риска, которые используются для выполнения критических функций, контактируют с центральной нервной системой или системой кровообращения или содержат вещества человеческого или животного происхождения (например, сердечные клапаны, неврологические имплантаты и рассасывающиеся швы).
В дополнение к этим общим правилам классификации существуют специальные правила для некоторых типов устройств, таких как противозачаточные устройства, дезинфицирующие средства и радиологические устройства.
Эти специальные правила могут привести к более высокой классификации, чем та, которая была бы присвоена только на основании общих правил.
Примеры классификаций устройств и их последствия для регистрации
Чтобы проиллюстрировать систему классификации и ее последствия для регистрации, рассмотрим следующие примеры:
- Нестерильные марлевые повязки (класс I): Нестерильные марлевые повязки относятся к классу I как устройства низкого риска, имеющие лишь кратковременный контакт с неповрежденной кожей. Производитель должен подготовить технический документ, демонстрирующий соответствие основным требованиям, но ему не нужно привлекать нотифицированный орган к процессу оценки соответствия. Регистрационное досье на устройство класса I относительно простое и может быть быстро обработано властями.
- Слуховые аппараты (Класс IIa): Слуховые аппараты классифицируются как устройства Класса IIa, поскольку они используются в отверстиях тела (ушной канал) и потенциально могут причинить вред в случае неисправности. Производитель должен подготовить более подробное техническое досье и привлечь нотифицированный орган к процессу оценки соответствия. Регистрационное досье на устройство класса IIa требует более подробной документации и может занимать больше времени, чем на устройство класса I.
- Ортопедические имплантаты (класс IIb): Ортопедические имплантаты, такие как протезы тазобедренного или коленного сустава, относятся к классу IIb из-за их длительной инвазивности и возможности получения серьезных травм в случае отказа. Производитель должен подготовить полное техническое досье, включая клинические данные, и пройти более строгую оценку соответствия со стороны нотифицированного органа. Регистрационное досье на устройство класса IIb является сложным и может потребовать дополнительной экспертной оценки со стороны властей, что приведет к увеличению времени обработки.
- Сердечные клапаны (Класс III): Сердечные клапаны относятся к устройствам Класса III, поскольку они имеют решающее значение для функционирования системы кровообращения и могут привести к серьезным последствиям в случае неисправности.
Производитель должен подготовить обширное техническое досье, включающее значительные клинические данные, и пройти тщательную оценку соответствия нотифицированным органом. Регистрационное досье для устройства класса III является очень сложным и может потребовать дополнительной экспертизы и консультаций с клиническими специалистами, что приводит к самым длительным срокам обработки среди всех классов устройств.
Правильно классифицировав свои устройства и подготовив соответствующую документацию, производители могут упростить процесс регистрации и избежать ненужных задержек или отказов.
Процедуры и сроки регистрации в Азербайджане
Чтобы зарегистрировать медицинское изделие в Азербайджане, международные менеджеры по регуляторным вопросам должны пройти соответствующую процедуру регистрации и представить полное досье в Центр аналитической экспертизы лекарственных средств.
Процесс регистрации состоит из двух основных процедур: общей процедуры регистрации и упрощенной процедуры регистрации.
Общая процедура регистрации медицинских изделий в Азербайджане
Общая процедура регистрации является стандартным способом регистрации медицинских изделий в Азербайджане. Эта процедура включает в себя тщательную оценку качества, безопасности и эффективности устройства Центром аналитической экспертизы лекарственных средств.
Ниже перечислены основные этапы и требования общей процедуры регистрации:
- Подача досье: Заявитель должен представить полное регистрационное досье в Центр аналитической экспертизы лекарственных средств. Досье должно включать следующие документы:
- Административные документы (например, форма заявления, доверенность, сертификаты)
- Техническая документация (например, описание устройства, назначение, информация о конструкции и производстве, анализ рисков, клиническая оценка)
- Маркировка и упаковочные материалы (например, инструкции по применению, этикетки, макеты).
- Досье должно быть представлено как в печатном, так и в электронном формате, вместе с требуемой регистрационной пошлиной. Административная часть досье должна быть на азербайджанском или русском языке, а техническая документация может быть на азербайджанском, русском или английском языках.
- Предварительное рассмотрение: После получения досье Центр аналитической экспертизы лекарственных средств проводит предварительную проверку, чтобы убедиться, что заявка является полной и соответствует формальным требованиям. Если будут выявлены какие-либо недостатки, заявитель будет уведомлен и ему будет предоставлен определенный срок для их устранения.
- Специализированная экспертиза: После того как досье проходит предварительную экспертизу, оно подвергается специализированной экспертизе, которая включает в себя детальную оценку качества, безопасности и производительности устройства. Этот этап включает в себя:
- оценку технической документации
- Лабораторный анализ образцов устройства, если требуется
- Анализ клинических данных и документации по управлению рисками
- На этом этапе Центр аналитической экспертизы лекарственных средств может запросить у заявителя дополнительную информацию или разъяснения. Очень важно быстро и подробно ответить на эти запросы, чтобы избежать задержек в процессе регистрации.
- Решение и выдача свидетельства: На основании результатов специализированной экспертизы Центр аналитической экспертизы лекарственных средств принимает решение об одобрении или отклонении заявки на регистрацию. В случае положительного решения заявитель получает регистрационное удостоверение, действительное в течение пяти лет. Зарегистрированное устройство также будет включено в национальный реестр медицинских изделий.
Общая процедура регистрации обычно занимает от 30 до 45 дней с момента подачи досье до выдачи регистрационного удостоверения.
Однако эти сроки могут варьироваться в зависимости от сложности устройства, качества поданного досье и загруженности Центра аналитической экспертизы лекарственных средств.
Упрощенная процедура регистрации медицинских изделий в Азербайджане
Азербайджан предлагает упрощенную процедуру регистрации медицинских изделий, которые уже были зарегистрированы в странах со строгими регулирующими органами или получили маркировку CE в соответствии с Положением Европейского Союза о медицинских изделиях (MDR).
Эта процедура направлена на упрощение процесса регистрации устройств, прошедших строгую оценку признанных регулирующих органов.
Чтобы получить право на упрощенную процедуру регистрации, медицинское изделие должно соответствовать одному из следующих критериев:
- Устройство зарегистрировано в странах с жесткими нормативными требованиями (например, в ЕС, США, Канаде, Японии, Австралии).
- Устройство получило маркировку CE в соответствии с Положением ЕС о медицинских изделиях (MDR).
В рамках упрощенной процедуры регистрации заявитель должен представить сокращенный набор документов, включая:
- Форму заявления и доверенность
- Свидетельство о регистрации или сертификат CE из страны регистрации
- Инструкция по применению и маркировочные материалы на азербайджанском языке
Центр аналитической экспертизы лекарственных средств рассмотрит представленные документы и примет решение о регистрации в течение 30 дней .
В случае положительного решения заявитель получит регистрационное удостоверение, действительное в течение того же срока, что и регистрационный или CE-сертификат из референтной страны, но не более пяти лет .
Важно отметить, что упрощенная процедура регистрации не освобождает устройства от требования продемонстрировать качество, безопасность и эффективность. Центр аналитической экспертизы лекарственных средств все равно может запросить дополнительную информацию или разъяснения в процессе рассмотрения.
Отказ в регистрации и процесс обжалования
В некоторых случаях Центр аналитической экспертизы лекарственных средств может отказать в регистрации медицинского изделия. Наиболее распространенные основания для отказа включают:
- недостаточные или неадекватные данные, подтверждающие качество, безопасность или эффективность устройства
- Несоблюдение соответствующих стандартов или нормативных требований
- Искажение или фальсификация информации в регистрационном досье
- неустранение недостатков или непредоставление дополнительной информации по запросу органов власти в установленные сроки.
В случае отказа в регистрации заявитель получает письменное уведомление с указанием причин отказа. Заявитель может обжаловать решение в течение 30 дней, подав письменное заявление в Министерство здравоохранения.
Апелляция будет рассмотрена экспертной комиссией более высокого уровня, которая примет окончательное решение по заявке на регистрацию.
Пострегистрационные обязательства в Азербайджане
После того как медицинское изделие было успешно зарегистрировано в Азербайджане, производители и уполномоченные представители должны соблюдать различные пострегистрационные обязательства, чтобы сохранить регистрацию устройства и обеспечить его безопасность и эффективность.
Требования к маркировке и упаковке в Азербайджане
Все медицинские изделия, продаваемые в Азербайджане, должны соответствовать определенным требованиям к маркировке и упаковке, чтобы медицинские работники и пациенты имели доступ к точной и полной информации об изделии.
Требования к языку и содержанию в Азербайджане
Маркировка и упаковочные материалы для медицинских изделий должны быть на азербайджанском языке.
На этикетке и в инструкции по применению должна быть указана следующая информация:
- Название и модель устройства
- Название и адрес производителя
- Назначение и показания к применению
- Противопоказания, предупреждения и меры предосторожности
- Инструкции по применению, хранению и утилизации
- Номер партии или серийный номер
- Срок годности или дата изготовления
- Статус стерильности и метод стерилизации, если применимо
- маркировка CE или регистрационный номер, если применимо
Помимо азербайджанского языка, маркировка и упаковочные материалы могут быть представлены на русском, английском или турецком языках.
Особые требования к некоторым категориям изделий
К некоторым категориям медицинских изделий могут предъявляться дополнительные требования к маркировке и упаковке. Например:
- Стерильные устройства должны содержать информацию о методе стерилизации и сроке действия стерильности
- Устройства, содержащие лекарственные вещества, должны содержать информацию о составе и дозировке вещества
- Устройства, излучающие ионизирующее излучение, должны содержать предупреждения и меры предосторожности, связанные с воздействием излучения.
Процесс утверждения изменений в маркировке и упаковке
Любые изменения в утвержденной маркировке или упаковочных материалах должны быть представлены в Центр аналитической экспертизы лекарственных средств для рассмотрения и утверждения перед внедрением.
Заявитель должен предоставить подробное описание предлагаемых изменений и их обоснование, а также обновленные образцы маркировки и упаковки.
Центр аналитической экспертизы лекарственных средств рассмотрит предложенные изменения и примет решение об их одобрении в течение 30 дней.
Если изменения будут признаны приемлемыми, заявитель получит уведомление об одобрении и сможет приступить к внедрению обновленной маркировки и упаковки.
Бдительность и отчетность о неблагоприятных событиях в Азербайджане
Производители и уполномоченные представители несут ответственность за постоянный мониторинг безопасности и эффективности своих медицинских изделий и сообщают о любых нежелательных явлениях или инцидентах в Центр аналитической экспертизы лекарственных средств.
Требования к созданию системы бдительности
Производители должны создать и поддерживать систему бдительности, соответствующую требованиям, изложенным в Законе о лекарственных средствах и соответствующих нормативных актах.
Ключевые элементы системы бдительности включают:
- Процедуры сбора, оценки и представления информации о неблагоприятных событиях и инцидентах
- Назначенное контактное лицо по вопросам бдительности, ответственное за связь с властями
- Ведение записей и документации по бдительности
- Проведение анализа тенденций и оценки рисков на основе данных о бдительности
Сроки и процедуры отчетности
Производители и уполномоченные представители должны сообщать в Центр аналитической экспертизы лекарственных средств о следующих типах событий:
- Серьезные нежелательные явления: в течение 15 календарных дней после того, как стало известно о событии
- Другие нежелательные явления и инциденты: в течение 30 календарных дней после того, как стало известно о событии.
- Корректирующие действия по обеспечению безопасности на местах (например, отзыв, модификации): до или одновременно с реализацией действия.
Отчеты должны быть представлены с использованием установленных форм и каналов, предоставленных Центром аналитической экспертизы лекарственных средств. Отчеты должны содержать подробное описание события, затронутого устройства (устройств) и действий, предпринятых или запланированных производителем для решения проблемы.
Сотрудничество с регулирующими органами в случае инцидентов или отзывов
В случае серьезного инцидента или необходимости проведения корректирующих мероприятий по обеспечению безопасности на местах производители и уполномоченные представители должны в полной мере сотрудничать с Центром аналитической экспертизы лекарственных средств и другими соответствующими органами.
Это сотрудничество может включать в себя:
- Предоставление дополнительной информации и документации по запросу
- Проведение расследований и анализ первопричин
- Осуществление корректирующих и профилактических действий
- Общение с медицинскими работниками и пациентами
- Содействие отзыву или модификации затронутых устройств.
Несоблюдение требований к бдительности и отчетности о неблагоприятных событиях может повлечь за собой наказание в виде штрафов, приостановки или отзыва регистрации устройства, а в тяжелых случаях — уголовную ответственность.
Постмаркетинговое наблюдение и клиническое сопровождение
Производители обязаны проводить постмаркетинговое наблюдение и клиническое сопровождение для обеспечения безопасности и эффективности медицинских изделий на протяжении всего их жизненного цикла.
Обязательства по проведению постмаркетингового наблюдения
Обязанности по проведению постмаркетингового наблюдения включают:
- сбор и анализ данных о безопасности и эффективности устройства из различных источников (например, отчетов о бдительности, жалоб потребителей, научной литературы)
- Проведение периодических обзоров безопасности и обновление документации по управлению рисками устройства
- Осуществление корректирующих и предупреждающих действий для решения выявленных проблем или рисков
- Ведение записей и документации по постмаркетинговому надзору
Периодические отчеты об обновлении информации о безопасности (PSURs)
Производители должны представлять периодические отчеты об обновлении безопасности (PSUR) в Центр аналитической экспертизы лекарственных средств в соответствии со следующим графиком:
- Устройства класса III: ежегодно
- Устройства класса IIb: каждые 2 года
- Устройства класса IIa: каждые 3 года
- Устройства класса I: не требуется
PSUR должны содержать всесторонний обзор безопасности и эффективности устройства, включая резюме данных о бдительности, мероприятиях по постмаркетинговому надзору, а также любые изменения или обновления в документации по управлению рисками устройства.
Клинические последующие исследования для устройств высокого риска в Азербайджане
Для устройств с высоким уровнем риска (например, класс III и некоторые устройства класса IIb) от производителей может потребоваться проведение постмаркетинговых клинических исследований для дальнейшей оценки безопасности и эффективности устройства в реальных условиях.
Необходимость проведения последующих клинических исследований будет определяться Центром аналитической экспертизы лекарственных средств в каждом конкретном случае с учетом таких факторов, как:
- новизна и сложность устройства
- Наличие и качество клинических данных, полученных до выхода устройства на рынок
- Потенциальные риски, связанные с использованием устройства
- Целевая популяция пациентов и предполагаемое использование.
Перед началом исследования производители должны представить протокол клинического повторного исследования в Центр аналитической экспертизы лекарственных средств для рассмотрения и утверждения.
Результаты исследования должны быть представлены в органы власти в установленные сроки и могут быть использованы для обновления маркировки устройства, документации по управлению рисками или регистрационного статуса.
Продление регистрации медицинских изделий и внесение изменений в регистрацию в Азербайджане
Регистрации медицинских изделий в Азербайджане действительны в течение ограниченного периода времени и должны продлеваться для сохранения разрешения на продажу изделия. Кроме того, любые существенные изменения или модификации зарегистрированного устройства должны быть доведены до сведения или одобрены Центром аналитической экспертизы лекарственных средств.
Срок действия регистрационных свидетельств на медицинские изделия
Регистрационные свидетельства на медицинские изделия в Азербайджане обычно действительны в течение пяти лет с даты выдачи. Однако для устройств, зарегистрированных по упрощенной процедуре регистрации на основании иностранной регистрации или сертификата CE, срок действия может быть короче и совпадать с датой истечения срока действия основной иностранной регистрации или сертификата CE.
Процесс подачи заявки на продление и требования
Чтобы продлить регистрацию медицинского изделия, производители или уполномоченные представители должны подать заявку на продление в Центр аналитической экспертизы лекарственных средств не позднее чем за 90 дней до истечения срока действия текущей регистрации.
Заявка на продление регистрации должна включать:
- обновленную техническую документацию, включая любые изменения или модификации, внесенные в устройство с момента первоначальной регистрации
- Актуальная маркировка и упаковочные материалы
- Данные бдительности и постмаркетингового наблюдения
- Доказательство оплаты пошлины за продление.
Центр аналитической экспертизы лекарственных средств рассмотрит заявку на продление и примет решение о продлении в течение 30 дней. Если заявка будет одобрена, производитель получит новое регистрационное удостоверение сроком действия еще на пять лет.
Уведомление и утверждение изменений в зарегистрированных медицинских изделиях
Производители должны уведомлять Центр аналитической экспертизы лекарственных средств о любых существенных изменениях или вариациях зарегистрированного медицинского изделия.
К типам изменений, требующих уведомления или одобрения, относятся:
- Изменения в конструкции, составе или процессе производства устройства
- Изменения в предназначении устройства
Правила импорта и экспорта в Азербайджане
Менеджеры по международным регуляторным вопросам также должны быть осведомлены о правилах импорта и экспорта медицинских изделий в Азербайджане, чтобы обеспечить их соблюдение и способствовать беспрепятственной трансграничной торговле.
Требования к лицензированию импортеров и экспортеров медицинских изделий
Компании, занимающиеся импортом или экспортом медицинских изделий в Азербайджане, должны получить соответствующие лицензии в Министерстве здравоохранения.
Лицензия импортера
Для импорта медицинских изделий в Азербайджан компании должны получить лицензию импортера. Основные требования для получения лицензии импортера включают:
- Регистрация юридического лица в Азербайджане
- Наличие соответствующих помещений и оборудования для хранения и обработки медицинских изделий
- наличие квалифицированного персонала, включая ответственного за контроль качества и соблюдение нормативных требований
- внедрение системы управления качеством, отвечающей соответствующим стандартам и нормам.
Заявление на получение лицензии импортера должно быть подано в Министерство здравоохранения вместе с подтверждающими документами, такими как регистрационное свидетельство компании, план помещений и квалификация персонала. Министерство рассмотрит заявку и примет решение в течение 30 дней.
Лицензированные импортеры несут ответственность за то, чтобы импортируемые медицинские изделия были зарегистрированы в Азербайджане, соответствовали действующим нормам, а также хранились и обрабатывались в соответствии с надлежащей практикой распространения.
Лицензия экспортера
Компании, экспортирующие медицинские изделия из Азербайджана, должны получить лицензию экспортера. Требования для получения лицензии экспортера аналогичны требованиям для получения лицензии импортера, с дополнительным требованием наличия действующей лицензии на производство или контракта с лицензированным производителем.
Процедура подачи заявки на получение лицензии экспортера также аналогична процедуре получения лицензии импортера: Министерство здравоохранения рассматривает заявку и принимает решение в течение 30 дней.
Лицензированные экспортеры несут ответственность за то, чтобы экспортируемые медицинские изделия соответствовали нормам страны назначения и сопровождались необходимой документацией, такой как сертификаты происхождения и соответствия.
Процедуры таможенного оформления и документация в Азербайджане
При импорте или экспорте медицинских изделий компании должны соблюдать процедуры таможенного оформления и предоставлять необходимую документацию.
Необходимые документы для импорта и экспорта
Для импорта и экспорта медицинских изделий в Азербайджан обычно требуются следующие документы:
- Товарная накладная
- Упаковочный лист
- Сертификат происхождения
- Сертификат соответствия или регистрации
- Грузовые документы (например, коносамент, авианакладная)
- Лицензия на импорт или экспорт
- Другие документы, требуемые таможенными органами.
Важно убедиться, что все документы точны, полны и при необходимости легализованы или апостилированы.
Сотрудничество с таможенными органами
Компании, импортирующие или экспортирующие медицинские изделия, должны сотрудничать с таможенными органами и предоставлять любую дополнительную информацию или документацию, запрашиваемую в процессе таможенного оформления.
Это может включать в себя:
- Предоставление образцов устройств для проверки или тестирования
- Уточнение предназначения или классификации устройств
- Разъяснение предназначения или классификации устройствДемонстрация соответствия действующим нормам и стандартам
Нежелание сотрудничать с таможенными органами или предоставлять требуемую документацию может привести к задержкам, штрафам или отказу в приеме груза.
Особые требования к устройствам, содержащим контролируемые вещества или требующим управления холодовой цепочкой
К медицинским изделиям, которые содержат контролируемые вещества (например, наркотики, психотропные вещества) или требуют управления холодовой цепочкой (например, чувствительные к температуре изделия), предъявляются дополнительные требования к импорту и экспорту.
Для импорта и экспорта устройств, содержащих контролируемые вещества, импортеры и экспортеры должны получить специальные разрешения Министерства здравоохранения и соблюдать правила обращения и транспортировки контролируемых веществ.
Для устройств, требующих управления «холодной цепью», импортеры и экспортеры должны обеспечить хранение и транспортировку устройств в соответствующих температурных условиях и предоставить доказательства мониторинга и контроля температуры при таможенном оформлении.
Рекомендации для международных компаний
Чтобы успешно ориентироваться на азербайджанском рынке медицинских изделий, менеджерам по международным регуляторным вопросам следует принять во внимание следующие рекомендации:
Важность привлечения местных экспертов и партнеров в области регулирования
Привлечение местных экспертов и партнеров, таких как Delta Medical, может оказать неоценимую помощь в понимании и соблюдении азербайджанских правил производства медицинских изделий. Местные эксперты могут помочь:
- Толкование и применение соответствующих законов и нормативных актов.
- Подготовка и подача регистрационных досье и других необходимых документов.
- Общение с регулирующими органами и ответы на запросы.
- Следите за изменениями в законодательстве и обновляйте стратегии соответствия.
При выборе местных партнеров международные компании должны учитывать такие факторы, как опыт, знания, репутация и способность партнера оказывать всестороннюю поддержку на протяжении всего жизненного цикла продукта.
Заключительные мысли о регистрации медицинских изделий в Азербайджане
Чтобы успешно пройти азербайджанские процедуры по медицинским изделиям, менеджерам по международным регуляторным вопросам следует принять во внимание следующие рекомендации:
- Привлекайте местных экспертов и партнеров по регулированию: Сотрудничество с местными экспертами и партнерами, такими как Delta Medical, может помочь международным компаниям понять и соблюсти азербайджанские требования, избежать распространенных «подводных камней» и ускорить процесс регистрации.
- Планируйте заранее и выделяйте достаточно времени на регистрацию: Процесс регистрации в Азербайджане может быть длительным и сложным, особенно для инновационных продуктов. Международным компаниям следует заранее планировать и выделять достаточно времени на подготовку, подачу и рассмотрение досье, чтобы избежать задержек с выходом на рынок.
- Приоритет продуктов с высоким рыночным потенциалом: Для получения максимальной отдачи от инвестиций международные компании должны определить приоритетность регистрации продуктов с высоким рыночным потенциалом в Азербайджане, учитывая такие факторы, как распространенность заболеваний, неудовлетворенные медицинские потребности и конкуренция.
- Обеспечьте соблюдение пострегистрационных требований: Международные компании должны создать надежные системы и процессы фармаконадзора, чтобы соответствовать пострегистрационным требованиям в Азербайджане. Регулярный пересмотр и обновление этих систем поможет избежать штрафов и обеспечить бесперебойную работу бизнеса.
- Будьте в курсе изменений и обновлений в нормативно-правовой базе: Нормативно-правовая база Азербайджана постоянно развивается, периодически вводятся новые правила и рекомендации. Международные менеджеры по регуляторным вопросам должны быть в курсе этих изменений и соответствующим образом адаптировать свои стратегии и процессы.
Следуя этим рекомендациям и тесно сотрудничая с местными партнерами, международные компании могут успешно работать на азербайджанском регуляторном рынке и внести свой вклад в улучшение доступа пациентов к безопасным, эффективным и высококачественным медицинским изделиям.


